Date: Mon, 22 Jan 2024 15:46:47 +0800 (GMT+08:00)
Dear AMBER Community,
I am currently conducting simulations of a protein-protein complex, employing restraints during the heating phase. However, I have observed that the protein-protein complex becomes more tightly packed upon transitioning from NVT to NPT.
While investigating this issue, I discovered a related inquiry on the AMBER mailing list (http://archive.ambermd.org/202102/0298.html), “by default, LEaP adds too little water so upon pressure scaling, the box has to shrink and therefore the restraint coordinates shrink.“
Unfortunately, I am facing challenges in implementing the solutions provided in the mailing list.
I have two specific questions for the community. Firstly, without additional intervention, would such a simulation be considered flawed? Secondly, are there more elegant methods to ensure correct scaling, perhaps through adjustable parameters?
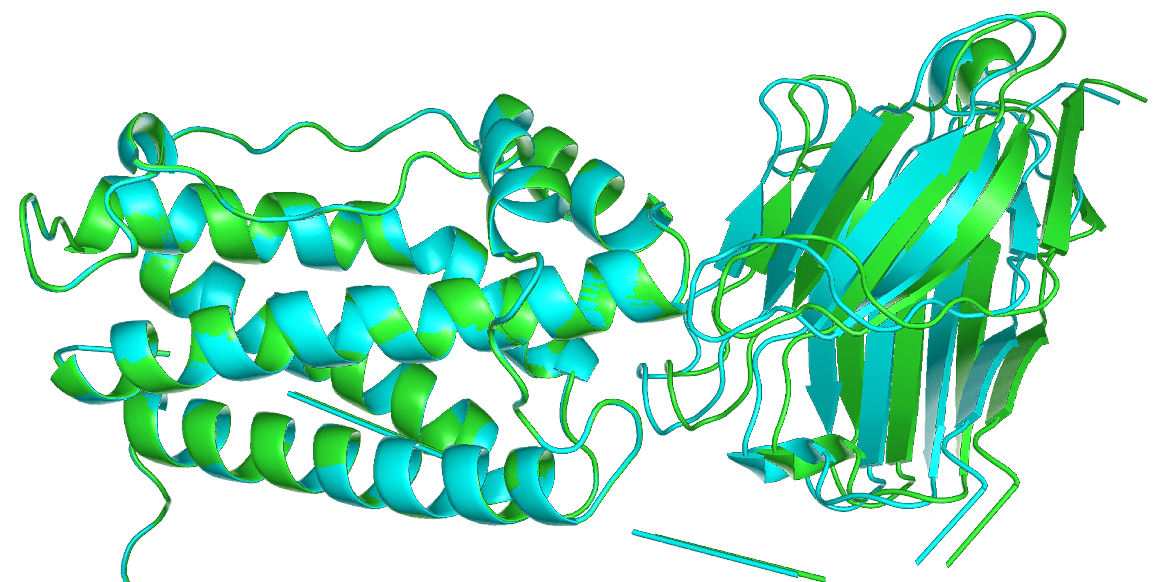
I have attached the parameter files (heat1 and heat2) and a structural comparison of the proteins for your reference. Any guidance or insights you can provide would be greatly appreciated.
Thank you for your time and support.
Best regards,
Xiaochun Zhang
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: heat1.in
- application/octet-stream attachment: heat2.in
(image/png attachment: antibody.png)
