From: David DELLEMME via AMBER <amber.ambermd.org>
Date: Fri, 22 Dec 2023 11:17:03 +0000
Hello Amber users,
I am trying to use the mdgx program to reparameterize peptoid molecules. I am only considering the dihedral parameters.
I tried to reproduce what was done for the Dreiding force-field, which was reparameterized against mp2/cc-pvdz QM data (
https://doi.org/10.1002/adts.201800089).
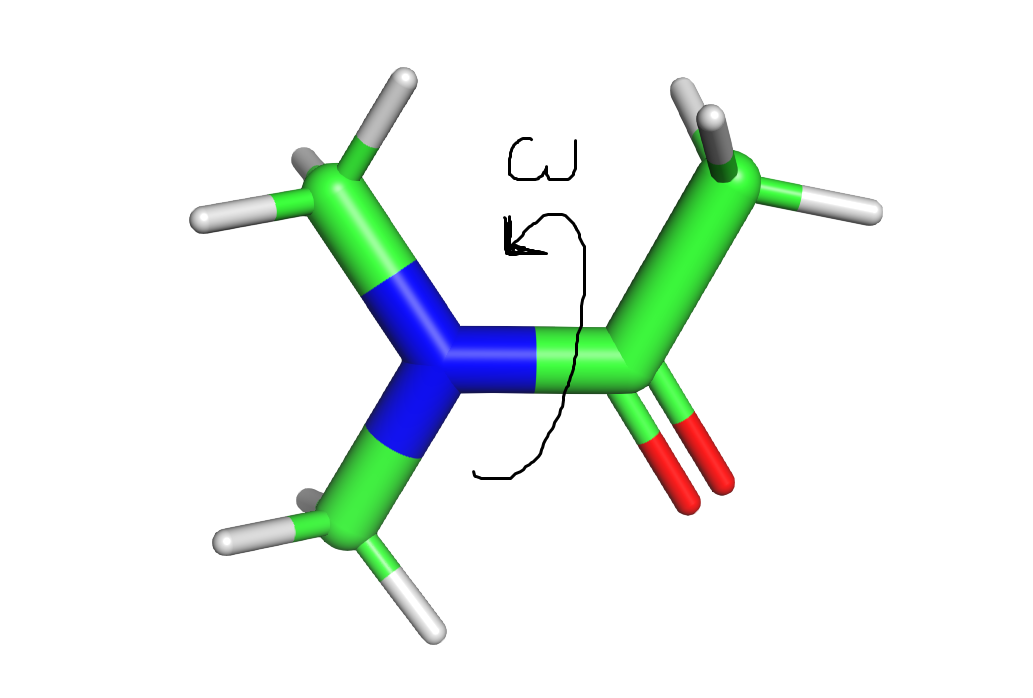
I generated conformers to sample the "omega" torsion, building the same fragment as in the "Pepdroid" methodology (see previous article). You can see the fragment and the torsion sampled in the beautiful picture attached. I generated conformers using the following input in mdgx to sample the omega torsion:
&configs
GridSample :1.C4 :1.N1 :1.C2 :1.C1 { -180.0 180.0 } Krst 64.0
count 360
verbose 1
qmlev 'MP2'
basis 'cc-pvDZ'
maxcore 2000
ncpu 1
chk checkpoint_file.dat
outbase 'backbone', 'backbone'
write 'cdf', 'gaussian'
outsuff 'nc', 'com'
maxcyc 3000
nshuffle 10
shuffle bootstrap
&end
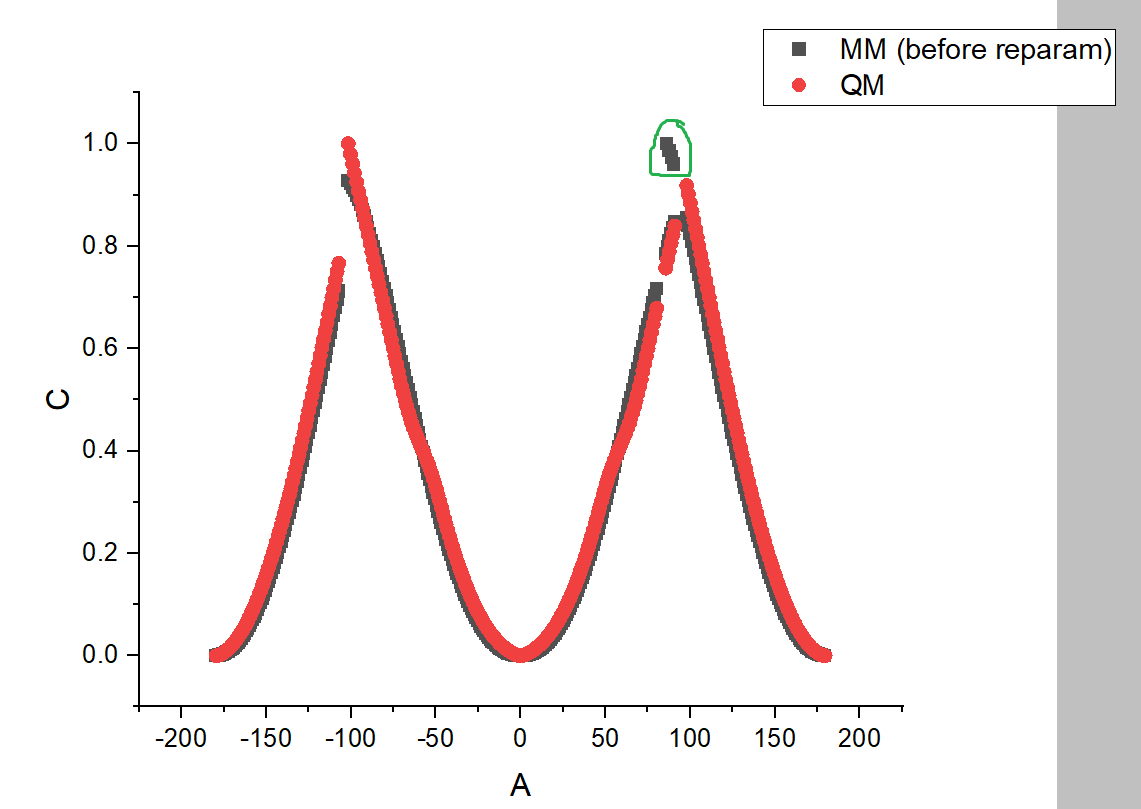
I also show you the torsional profile that I get for the generated conformers, with relative energy in Y and dihedral angle in X (the 4 points circled in green have not been used for QM calculations and further reparameterization, as they clearly seemed to be out of the "normal" torsional profile, which is why the points in the QM curve are missing). I find this curve a bit weird, it looks like all the points are not on the same curve. Could it be that all the conformations are not global minima, but trapped local minima? If yes, how to avoid them? Even using the "nshuffle" option to restart the optimization step does not help to avoid the fact that some points seem "out of the curve".
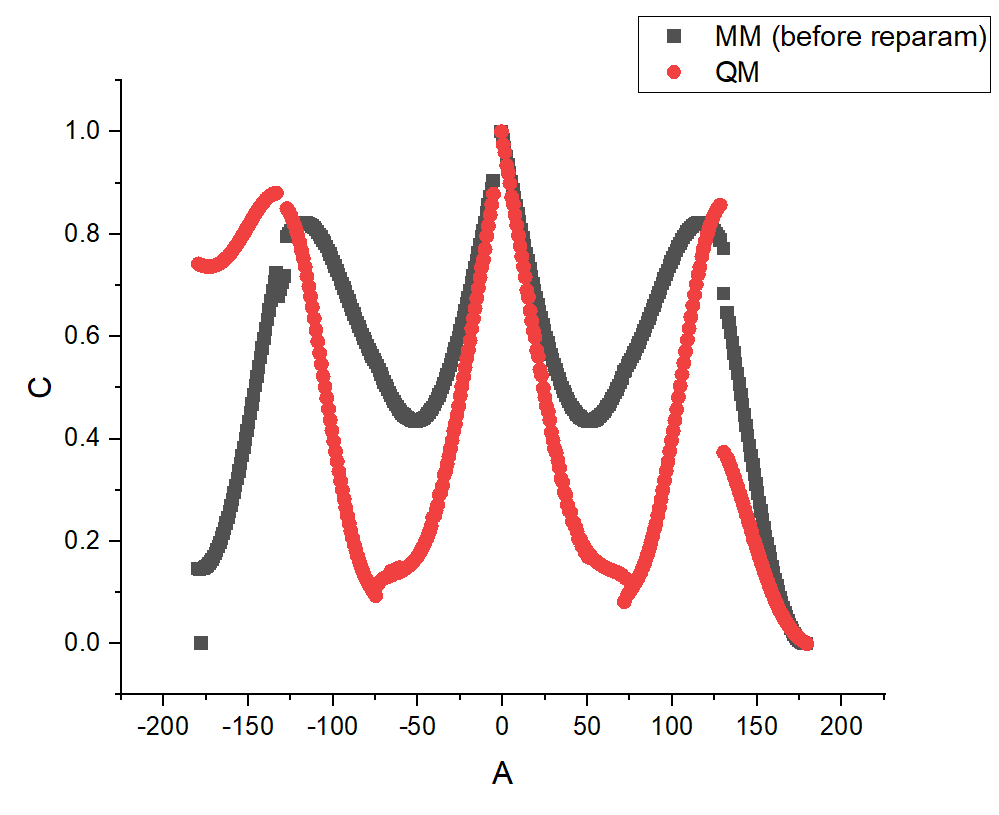
Of course, this example is not so bad, the maxima/minima are found, but I wonder why the curve has this weird shape. And this is a problem because, in the reparameterization of other torsional angles, the curves are much worse (see an example in the third picture attached). Also, the fragments used to do the reparameterizations are the same than in the "Pepdroid" paper and the charges were attributed using the RESP methodology.
So, I am a bit stuck here and would like to know if any of you would have suggestions on how to improve my methodology, or if it is usual to have such "weird points"?
Thank you very much in advance for your help, and do not hesitate if you need further information!
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Fri Dec 22 2023 - 03:30:02 PST