Dear Amber community,
Referring to tutorial Amber tutorial - Protein Ligand binding free energies (ambermd.org), I'm trying to run the QM/MM simulation to collect potential energy differences for FEP calculations. The version I am using is Amber22. The simulation process is calculated using Sander.
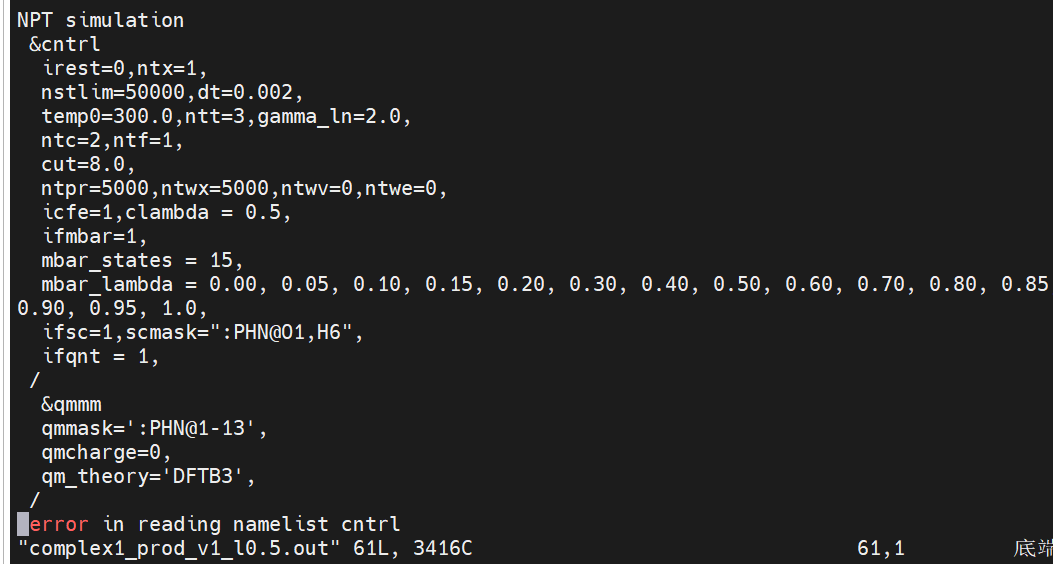
When I generated an input file using the new version of the mbar method in the Amber22 manual, an error was reported('error in reading namelist cntrl').
Here is the input file:
NPT simulation
&cntrl
irest=0,ntx=1,
nstlim=50000,dt=0.002,
temp0=300.0,ntt=3,gamma_ln=2.0,
ntc=2,ntf=1,
cut=8.0,
ntpr=5000,ntwx=5000,ntwv=0,ntwe=0,
icfe=1,clambda = 0.5,
ifmbar=1,
mbar_states = 15,
mbar_lambda = 0.00, 0.05, 0.10, 0.15, 0.20, 0.30, 0.40, 0.50, 0.60, 0.70, 0.80, 0.85, 0.90, 0.95, 1.0,
ifsc=1,scmask=":PHN.O1,H6",
ifqnt = 1,
/
&qmmm
qmmask=':PHN.1-13',
qmcharge=0,
qm_theory='DFTB3',
/
And my terminal's command is ' mpirun -np 2 sander.MPI -ng 2 -groupfile group_prod_l ' .
When I generate an input file using the old version of Amber method, it can be run. The input file is as follows:
NPT simulation
&cntrl
irest=0,ntx=1,
nstlim=500000,dt=0.001,
temp0=300.0,ntt=3,gamma_ln=2.0,
ntc=2,ntf=1,
cut=8.0,
ntpr=5000,ntwx=5000,ntwv=0,ntwe=0,
icfe=1,clambda = 0.5,
ifmbar=1, bar_intervall=1000,bar_l_min=0.005,bar_l_max=0.995,bar_l_incr=0.1,
ifsc=1,scmask=":BNZ.H6",
ifqnt = 1,
/
&qmmm
qmmask=':BNZ.1-12',
qmcharge=0,
qm_theory='DFTB3',
/
However, in the old version of Amber method, it is not allowed for bar_l_min>0.995 or bar_l_max<0.005, which makes it impossible for me to set the lambda=0,1.
How should I do?
Best wishes!
Leveen
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Sat Aug 05 2023 - 18:00:02 PDT
{kind=link}
