Date: Thu, 3 Aug 2023 13:46:26 -0700
Hi Adrian,
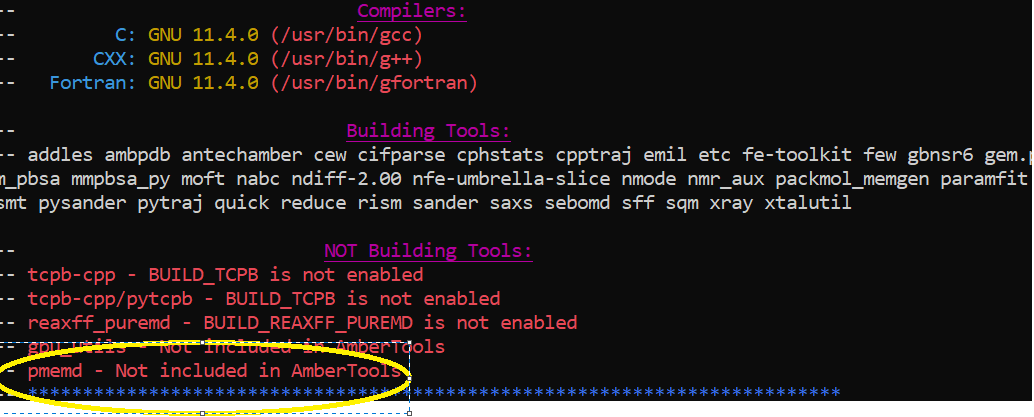
For the installation, I followed the instructions on the website and
downloaded the AmberTools23.tar file. When I ran "run_cmake" command, after
a while most libraries and tools were built, however, pmemd is not built as
it is not included in AmberTools. Therefore, later on during the tutorial I
ran into the previous issues if I want to replace gm.pmemd with pmemd. What
should I do then?
[image: image.png]
Maryam
On Thu, Aug 3, 2023 at 10:01 AM Adrian Roitberg via AMBER <amber.ambermd.org>
wrote:
> Maryam
>
> Are you trying to use GEM ?
>
> If not, then you must use pmemd, not gem.pmemd
>
> If pmemd is not in your bin directory, then that is your problem. Either
> it did not get compiled at all, or it tried and did not succeed.
>
> Focus on that first, how to get pmemd compiled (or pmemd.cuda if you are
> using GPU)
>
> Adrian
>
>
> On 8/3/23 12:55 PM, Maryam Foroozmehr via AMBER wrote:
> > [External Email]
> >
> > To Whom It May Concern:
> >
> > It is very appreciated letting me know how to debug the below problem.
> > Thank you.
> >
> > Simple simulation of Alanine dipeptide:
> > *Part 19. Run the production MD of alanine dipeptide with pmemd:*
> >
> > Question: There is no "pmemd" in the bin file, instead there is
> > "gem.pmemd". While running it there is the below error:
> >
> > ERROR: SHAKE (ntc != 1) may not be used with Amoeba or GEM!
> >
> > *- By considering the error, I made these changes: ntc=1,ntf=1, but then
> I
> > got the below error:*
> > *Command:* amber22_src/AmberTools/src/gem.pmemd/src/inpcrd_dat.F90 (unit
> =
> > 9, file = '02_Heat.ncrst')
> > Fortran runtime error: Bad value during floating point read
> >
> > Error termination. Backtrace:
> > #0 0x7f3684ffd960 in ???
> > #1 0x7f3684ffe4d9 in ???
> > #2 0x7f368524faba in ???
> > #3 0x7f36852536b9 in ???
> > #4 0x7f3685254e55 in ???
> > #5 0x559ec62a84f3 in __inpcrd_dat_mod_MOD_init_old_type_inpcrd_dat
> > #6 0x559ec62a9d8f in __master_setup_mod_MOD_master_setup
> > #7 0x559ec62d646f in MAIN__
> > #8 0x559ec61e858e in main
> > ^C
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
>
> --
> Dr. Adrian E. Roitberg
> V.T. and Louise Jackson Professor in Chemistry
> Department of Chemistry
> University of Florida
> roitberg.ufl.edu
> 352-392-6972
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
