Date: Fri, 23 Jun 2023 18:35:42 +0800 (GMT+08:00)
Hi all,
I encountered a problem when using the keyword ntwf = -1, as I would like to compute the forces acting on each atom, I then followed the instruction from the manual..
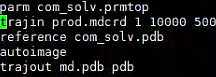
However, when I would like to convert the mdcrd file to the .pdb format using cpptraj with the .in file (as shown below), it is not possible to obtain the forces acting on each atom.
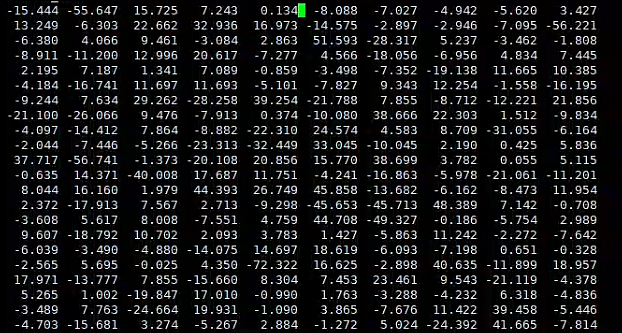
Also, I have also tried to set ntwf = 500 intended to obtain the mdfrc file with using ioutfm = 0 to get the ACSII format (as shown below), but I have difficulty in finding what is the meaning of the corresponding column. Could you please also give me some instructions ?
Thank you very much for your time, and please let me know if I can provide you more information to have your instructions.
Regards,
Kelvin
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber

(image/png attachment: 1687514909956.png)
(image/png attachment: 1687515048052.png)
(image/png attachment: 1687515158954.png)
