Date: Sun, 5 Mar 2023 06:42:26 +0000
Hello Amber Team
I have installed AMBER22 on the Cray EX system by using PrgEnv-gnu/8.3.3, and this is my run_cmake
cmake $AMBER_PREFIX/amber22_src \
-DCMAKE_INSTALL_PREFIX=$AMBER_PREFIX/amber22 \
-DCOMPILER=MANUAL \
-DCMAKE_C_COMPILER=cc -DCMAKE_CXX_COMPILER=CC -DCMAKE_Fortran_COMPILER=ftn \
-DMPI_C_COMPILER=$CRAY_MPICH_DIR/bin/mpicc \
-DMPI_CXX_COMPILER=$CRAY_MPICH_DIR/bin/mpicxx \
-DMPI_Fortran_COMPILER=$CRAY_MPICH_DIR/bin/mpifort \
-DMPI=TRUE -DCUDA=TRUE -DINSTALL_TESTS=TRUE \
-DCUDA_TOOLKIT_ROOT_DIR=/opt/nvidia/hpc_sdk/Linux_x86_64/21.9/cuda/ \
-DCMAKE_C_FLAGS="-L/opt/cray/pe/mpich/8.1.17/gtl/lib -lmpi_gtl_cuda" \
-DCMAKE_CXX_FLAGS="-L/opt/cray/pe/mpich/8.1.17/gtl/lib -lmpi_gtl_cuda" \
-DCMAKE_Fortran_FLAGS="-L/opt/cray/pe/mpich/8.1.17/gtl/lib -lmpi_gtl_cuda" \
-DCUDA_NVCC_FLAGS="-L/opt/cray/pe/mpich/8.1.17/gtl/lib -lmpi_gtl_cuda -Xcompiler -fpic" \
-DCMAKE_PREFIX_PATH=/opt/nvidia/hpc_sdk/Linux_x86_64/21.9/math_libs/11.4/lib64/ \
-DDOWNLOAD_MINICONDA=TRUE \
2>&1 | tee cmake.log
The result is that I can run serial and MPI jobs with no serious problem. However, when I run on GPU, I can only request 1 MPI task via SLURM. If I request more than 1 task, the error is shown in slurm output file:
cudaIpcOpenMemHandle failed on gpu->pbPeerAccumulator Handle invalid argument
cudaIpcOpenMemHandle failed on gpu->pbPeerAccumulator Handle invalid argument
srun: error: lanta-g-001: tasks 0-1: Exited with exit code 255
cudaIpcOpenMemHandle failed on gpu->pbPeerAccumulator Handle invalid argument
cudaIpcOpenMemHandle failed on gpu->pbPeerAccumulator Handle invalid argument
srun: error: lanta-g-001: tasks 0-1: Exited with exit code 255
cudaIpcOpenMemHandle failed on gpu->pbPeerAccumulator Handle invalid argument
cudaIpcOpenMemHandle failed on gpu->pbPeerAccumulator Handle invalid argument
srun: error: lanta-g-001: tasks 0-1: Exited with exit code 255
There is no problem if I request for 1 task and 1 GPUs card (AMBER still use 1 GPUs card when asking for more than 1)
Here is another info that could be useful
This one :
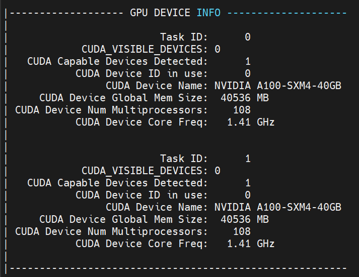
#SBATCH -N 1 --ntasks-per-node=2
#SBATCH --gpus-per-task=1
[cid:720e9b78-23ea-4e56-b8a2-88d63254acda]
and this:
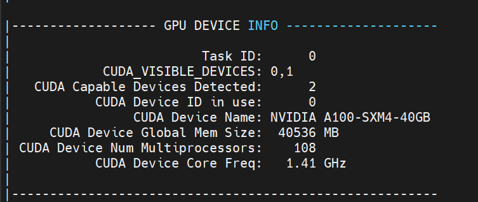
#SBATCH -N 1 --ntasks-per-node=1
#SBATCH --gpus-per-task=2
[cid:fbc938ce-488f-4e60-ac60-07692d91de4a]
I am the beginner, but it seems odd that CUDA Capable Devices is detected: 2 when request only 1 task. On the other hand, it can see only 1 CUDA Capable Devices when requesting for more than 1 task.
Do you have any suggestion for this error?
________________________________
Disclaimer:
This e-mail and any files transmitted with it may contain confidential and proprietary information of the National Science and Technology Development Agency (NSTDA), Thailand. They are intended solely for the use of the addressed individuals or entities. If you are not the intended recipient, you are required to immediately delete this e-mail and its contents from your system. Any disclosure, distribution, or action based upon the contents of this e-mail is strictly prohibited. Any views or opinions presented in this e-mail are solely those of the sender and do not necessarily represent those of NSTDA. NSTDA does not accept any responsibility for the content of this message or the consequences of any actions taken on the basis of the information provided. NSTDA accepts no liability for any damage caused by any virus or malware which may be inserted in this e-mail during transmission.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: GPU_info.png)
(image/png attachment: GPU_info2.png)
