Date: Thu, 16 Feb 2023 14:33:35 +0000
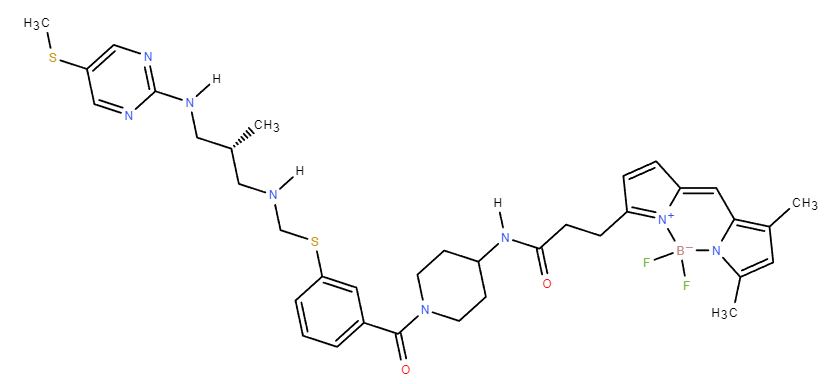
[cid:bb0b93dd-7d06-4f09-adb4-264bc2325a80]
above is the structure of the drug. I added hydrogen atoms in chimera. At the end of the step, chimera pointed out that the B atom needs assignment. I chose the geometry of B as tetragonal and with no subsitution. I am not sure if that is correct. Then when I added charge (neutralize it), it did not work. Maybe that is the reason why if failed? Could I know what kind of assignment on geometry and substitution I should use? Thanks a lot.
Laura
________________________________
From: Steinbrecher, Thomas <thomas.steinbrecher.roche.com>
Sent: Thursday, February 16, 2023 2:56
To: laura zhang <lqzhang0332.hotmail.com>; AMBER Mailing List <amber.ambermd.org>
Subject: Re: [Sender Not Verified] [AMBER] drug including B atom
Hi Laura,
it looks like your input structure has an issue. The total number of electrons comes out as odd so you are likely missing either a hydrogen or a charge somewhere (I'd also have a look at those long bond warnings). Maybe your B-atom is engaged in an odd bonding arrangement as boron is wont to do? Without seeing the structure it is hard to tell, but try and visualize it and check if you have a chemically reasonable structure with all valences saturated.
Kind Regards,
Thomas
On Thu, Feb 16, 2023 at 3:49 AM laura zhang via AMBER <amber.ambermd.org<mailto:amber.ambermd.org>> wrote:
Dear amber users,
I am working on a drug molecule which has a B atom. I got the following error message when trying to generate forcefield parameters for this molecule using antechamber.
-- Check Geometry --
for those bonded
Warning: Large distance for BOND 80 C27 F 0 1.54 [0.90 - 1.54]
Warning: Large distance for BOND 81 C27 F1 0 1.54 [0.90 - 1.54]
for those not bonded
-- Check Weird Bonds --
Status: pass
-- Check Number of Units --
Status: pass
acdoctor mode has completed checking the input file.
Info: Bond types are assigned for valence state (7) with penalty (1).
Info: Total number of electrons: 393; net charge: 0
Info: The number of electrons is odd (393).
Please check the total charge (-nc flag) and spin multiplicity (-m flag).
Running: /home/amber20/bin/sqm -O -i sqm.in<https://na01.safelinks.protection.outlook.com/?url=http%3A%2F%2Fsqm.in%2F&data=05%7C01%7C%7Cafde1dabf26942afff4f08db0ff35819%7C84df9e7fe9f640afb435aaaaaaaaaaaa%7C1%7C0%7C638121310049345721%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=ZHjw1m8rVKqLpwvWHXS8QR3KKdKbx7cVqtqRjNFANbo%3D&reserved=0> -o sqm.out
/home/amber20/bin/wrapped_progs/antechamber: Fatal Error!
Cannot properly run "/home/amber20/bin/sqm -O -i sqm.in<https://na01.safelinks.protection.outlook.com/?url=http%3A%2F%2Fsqm.in%2F&data=05%7C01%7C%7Cafde1dabf26942afff4f08db0ff35819%7C84df9e7fe9f640afb435aaaaaaaaaaaa%7C1%7C0%7C638121310049345721%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=ZHjw1m8rVKqLpwvWHXS8QR3KKdKbx7cVqtqRjNFANbo%3D&reserved=0> -o sqm.out".
Cannot open file (drug_antechamber.mol2) with mode (r).
No such file or directory
I am wondering how to solve the issue. Thank you very much.
Laura
_______________________________________________
AMBER mailing list
AMBER.ambermd.org<mailto:AMBER.ambermd.org>
http://lists.ambermd.org/mailman/listinfo/amber<https://na01.safelinks.protection.outlook.com/?url=http%3A%2F%2Flists.ambermd.org%2Fmailman%2Flistinfo%2Famber&data=05%7C01%7C%7Cafde1dabf26942afff4f08db0ff35819%7C84df9e7fe9f640afb435aaaaaaaaaaaa%7C1%7C0%7C638121310049345721%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=xG0Gq7msQPUQmkiSUg9odscjamP3yl4B7nx5zaY%2Bqh0%3D&reserved=0>
-- Dr. Thomas Steinbrecher Principal Scientist CADD Roche Pharma Research and Early Development Roche Innovation Center Basel F. Hoffmann-La Roche Ltd Bldg. 092/3.92 Grenzacherstrasse 124 4070 Basel Switzerland Phone +41 61 682 1319 mailto: thomas.steinbrecher.roche.com<mailto:thomas.steinbrecher.roche.com>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
