Date: Thu, 27 Oct 2022 17:11:05 +0300
Dear all,
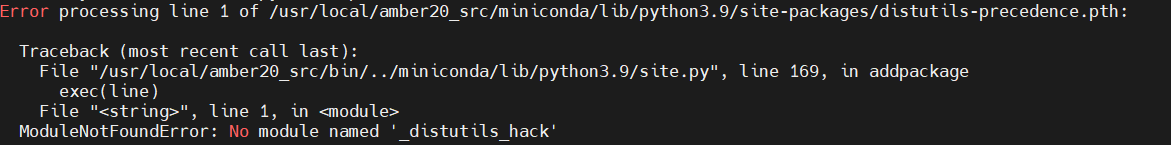
Is it normal to get such a warning every time when I run MCPB.py?
Thank you!
Best
Işılay
[image: image.png]
ışılay öztürk <isilayozturk.gmail.com>, 27 Eki 2022 Per, 15:03 tarihinde
şunu yazdı:
> ok, that is reasonable but I tried with gaussian 16 but I will check it.
>
> Thank u!
>
> Best regards
>
> Işılay
>
>
> Barış KURT <peacewolfus09.gmail.com>, 27 Eki 2022 Per, 14:59 tarihinde
> şunu yazdı:
>
>> Dear isilay,
>> it may beacuse of the your GAMESS-US version (and some old gaussian
>> versions also may lead the same problem) . To change version of it may work.
>> Good Luck,
>> Baris KURT
>> MSU TURKEY
>>
>> ışılay öztürk via AMBER <amber.ambermd.org>, 27 Eki 2022 Per, 14:33
>> tarihinde şunu yazdı:
>>
>>> Dear Yeler,
>>> I managed the that one but now I have new error. and I have no idea about
>>> that.
>>>
>>> https://ambermd.org/tutorials/advanced/tutorial20/mcpbpy_heme.htm
>>> Herein we use the ChgModB to perform the RESP charge fitting and generate
>>> the mol2 files for the metal site residues. Other options are also
>>> available: ChgModA, ChgModC and ChgModD (as 3a, 3c and 3d respectively).
>>> MCPB.py -i 4ZF6.in -s 3
>>>
>>> I'm trying to apply this step (for my system) in the tutorial, but I
>>> can't
>>> understand what the error is caused by. Do u have any idea about that.
>>> Thank u in advance and thank you for your reply.
>>>
>>> Best regards
>>>
>>> Işılay
>>>
>>> [image: image.png]
>>>
>>> Erdem Yeler <erdemyeler.gmail.com>, 27 Eki 2022 Per, 12:30 tarihinde
>>> şunu
>>> yazdı:
>>>
>>> > you said your ligand is:2naa_mol2files but error says: naa_mol2files
>>> not
>>> > found. Could you correct your file name and try again please?
>>> >
>>> >
>>> > ışılay öztürk via AMBER <amber.ambermd.org>, 13 Eki 2022 Per, 00:13
>>> > tarihinde şunu yazdı:
>>> >
>>> >> Dear all,
>>> >> Dear all,
>>> >>
>>> >> I have some problem about using *MCPB.py. *
>>> >> https://ambermd.org/tutorials/advanced/tutorial20/mcpbpy_heme.htm
>>> >> https://ambermd.org/tutorials/advanced/tutorial20/mcpbpy.htm
>>> >>
>>> >> I am trying to apply these tutorials to my structure. but It is always
>>> >> giving to this error: Traceback (most recent call last):
>>> >> File "/usr/local/amber20/bin/MCPB.py", line 581, in <module>
>>> >> raise pymsmtError('%s is required in naa_mol2files but not '
>>> >> pymsmt.exp.pymsmtError: LIG is required in naa_mol2files but not
>>> provided.
>>> >>
>>> >> If I am not wrong "2naa_mol2files " is defining to my ligand part.
>>> >>
>>> >> So How can I fix this problem?
>>> >>
>>> >> Thank you in advance
>>> >>
>>> >> Best,
>>> >>
>>> >> Işılay
>>> >> _______________________________________________
>>> >> AMBER mailing list
>>> >> AMBER.ambermd.org
>>> >> http://lists.ambermd.org/mailman/listinfo/amber
>>> >>
>>> >
>>> _______________________________________________
>>> AMBER mailing list
>>> AMBER.ambermd.org
>>> http://lists.ambermd.org/mailman/listinfo/amber
>>>
>>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
