Date: Sat, 2 Jul 2022 20:27:40 +0800 (GMT+08:00)
Dear Users,
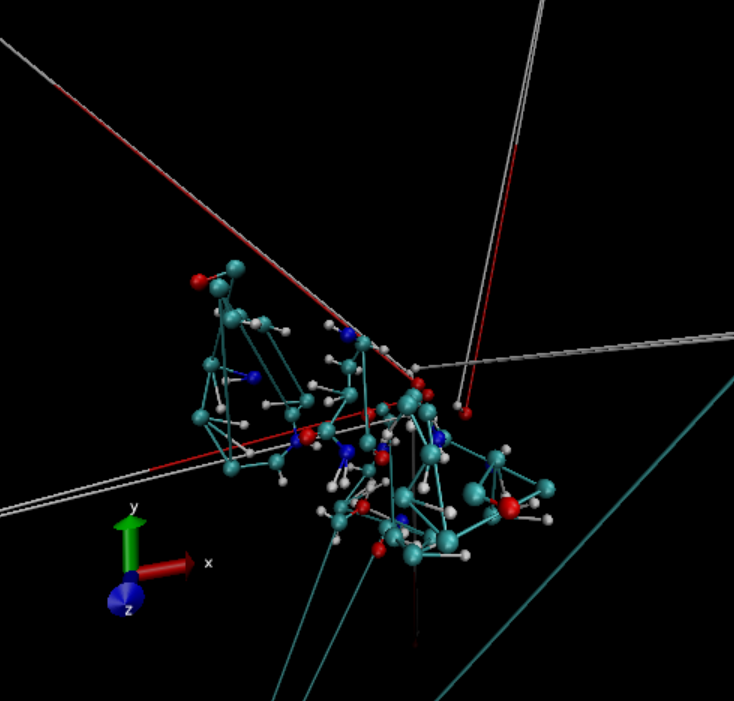
I am runing an equilibration with CPU version pmemd. However, I found the atoms are messy in the trajectory (see the figure 1 below). I tested it with GPU version. The trajectory is normal. Besides, I also tested another system and same problem was found: the trajectory is normal for GPU, yet not for CPU. The input parameters was attached in Figure 2. Is it the problem of the input parameters?
Thanks for your help in advance.
Figure 1.
Figure 2.
Zhanfeng Wang
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 1656764305243.png)

(image/png attachment: 1656764789897.png)
