Date: Fri, 8 Apr 2022 10:10:23 -0400
Hi,
Thanks for the files you sent.
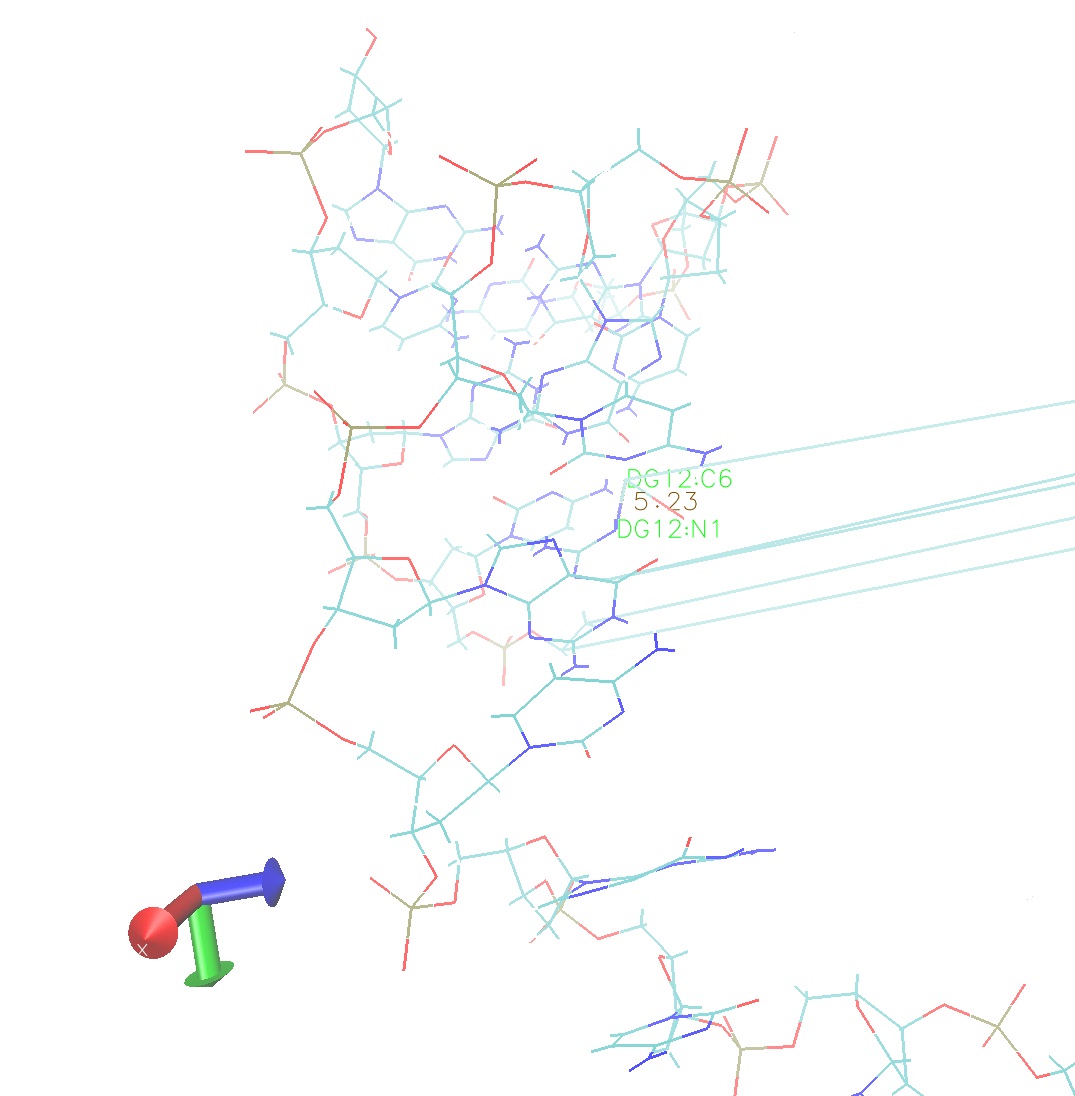
The problem here isn't imaging; something is definitely wrong with
your trajectory; as you can see from pstep_198.nc, frame 428 (the one
I mentioned before), not only do you have the strange artifact where
part of a DNA strand has apparently been imaged leading to distorted
bonds, parts of the structure which are not imaged are distorted (I've
highlighted a distorted bond in the attached picture).
I have a theory as to what went wrong based on the run script you've
sent me. You have the following in your script:
mpirun -np 2 pmemd.cuda -O -i mdp.inp -o pstep_${itr}.out -r
pstep_${itr}.rst -x pstep_${itr}.nc -p ../input/prmtop -c
../equilibration/step10.rst
You're using mpirun to run the *serial* program pmemd.cuda, i.e. you
are running two single separate instances of pmemd.cuda that are
competing for the same output files. I'm guessing this is what led to
the weird artifacts in your simulations. If you want to run in
parallel you would need to use pmemd.cuda.MPI. However, note that
speedup across GPUs is generally not efficient; you are encouraged to
benchmark and see if it really improves things that much. If you have
two GPUs it's probably better to run two separate simulations for
better sampling.
-Dan
On Wed, Apr 6, 2022 at 9:47 AM Daniel Roe <daniel.r.roe.gmail.com> wrote:
>
> Hi,
>
> What version of cpptraj are you using? I recently fixed an issue that
> could impact imaging in cases where there is non-contiguous atom
> ordering. You may want to try the latest version of cpptraj and see if
> 'autoimage' works.
>
> Otherwise feel free to send me your topology and coordinates off list
> so I can try to reproduce your issue.
>
> -Dan
>
> On Wed, Apr 6, 2022 at 4:07 AM Sathyaseelan C <bo17resch11006.iith.ac.in> wrote:
> >
> > Dear Amber users,
> >
> >
> >
> > I am facing an imaging issue during the MD simulation of a
> > DNA duplex with trivalent cations. The maging issue happens throughout the
> > simulation due to the terminal dimer of one of the DNA strands as well as
> > some of the trivalent ions. For instance, in one step the DNA dimer is out
> > of the box (as a fragment), but, in the next step it comes back to the
> > periodic box and becomes part of the duplex. This happens throughout the
> > simulation. I tried with imaging options as below, but, were not useful.
> > Any suggestion is highly appreciated.
> >
> >
> >
> >
> > *For RMSD*
> >
> >
> >
> > *1) **Using Image option*
> >
> > parm prmtop
> >
> > trajin trajin.nc
> >
> > reference ../../input/prmcrd
> >
> > center origin :1-32 # DNA residues
> >
> > image origin center
> >
> > center origin :33-44 # trivalent cations
> >
> > image origin center
> >
> > strip :WAT
> >
> > strip :LA
> >
> > strip :CL
> >
> > trajout test.pdb pdb
> >
> > go
> >
> >
> >
> > *2) **Using Autoimage option*
> >
> >
> >
> > parm prmtop
> >
> > trajin trajin.nc
> >
> > reference ../../input/prmcrd
> >
> > autoimage
> >
> > strip :WAT
> >
> > strip :LA
> >
> > strip :CL
> >
> > trajout test.pdb pdb
> >
> > go
> >
> >
> >
> >
> >
> >
> >
> > Thanks in advance
> >
> > Sathyaseelan
> >
> > --
> > *Thanks & Regards*
> >
> > *C .Sathyaseelan *
> > *PhD Research Scholar*
> > *C/o Dr. Thenmalarchelvi Rathinavelan*
> > *Molecular Biophysics Lab*
> > *Dept. of Biotechnology*
> > *Indian Institute of Technology (IIT)*
> > *Kandi, Hyderabad, Telangana - 502285*
> > *Ph.No 6383836804, 8300150807*
> >
> > --
> >
> >
> > Disclaimer:- This footer text is to convey that this email is sent by one
> > of the users of IITH. So, do not mark it as SPAM.
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: vmd.weirdbond.2.jpg)
