Date: Fri, 1 Apr 2022 12:17:04 +0300
Hello amberers,
I parameterized Pd atoms with almost all known bonds. For this, I wrote a
script to combine gaff and a novel metal force-field (the script can be
adapted with Pt, Au, Se, exc...) Everything works perfectly, but there is a
mini problem: Sander minimized the structure with very high accuracy, but Pd
lost its bonds and seemed like a "P" atom when I visualized it (I changed
the "Pd" sign to "Pm" (because in gaff there is pd even if it it is not
exactly Pd with capital P) but I couldn't solve the problem.) Why is this
happening? Actually, I don't see it as a serious problem because the
minimized geometry is correct, but if there is a way to get rid of this, I
am ready to apply.
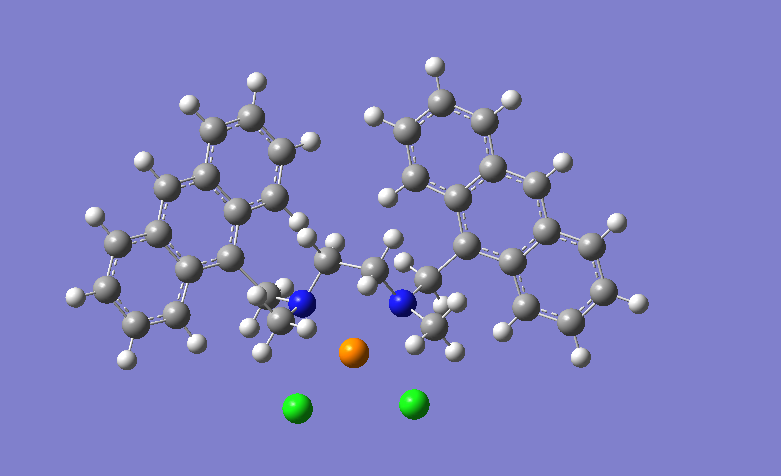
Here is the picture of my molecule: P must be Pd and must have bonds with
Cl and N atoms. I checked the prmtop file with parmed. PArmed seed Pd atom
and related bonds but after ambpdb commands it seems bond-less P atom...
[image: image.png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
