Date: Sun, 5 Sep 2021 21:39:05 +0800 (CST)
Hi, David,
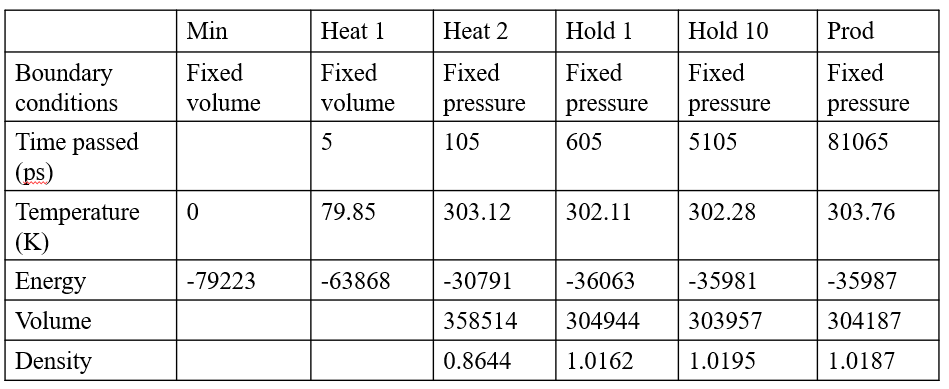
Thanks for your concern. The last email has some problems of layout. Here is the summarized table of MD result.
I am not familiar with how to analyze the trajectory file by cpptraj. So I extract some data from output file.
I don't know why there are larger density and smaller volume recorded after 600 ps than those at 105 ps. I see the systems dramatically enlarging after 105 ps (hold and production).
Should I change the boundary condition to fix the volume instead of the pressure at these two stage?
And I also concern that I sourced ff14SB force field instead of ff12SB that used in tutorial web since I did not find it. Does this significantly influence the result?
Best regards,
Wang Wei
At 2021-09-04 19:57:34, "David A Case" <dacase1.gmail.com> wrote:
>On Sat, Sep 04, 2021, wangwei1669 wrote:
>>
>>When I was following the production step of tutorial 1.7 "An Amber Lipid
>>Force Field Tutorial: Lipid14", I encountered a error about periodic box
>>dimensions.
>>
>>"Lipid production 303K 125ns
>> ntb=2, ! Constant pressure periodic boundary conditions
>> ntp=2, ! Anisotropic pressure coupling
>>The simulation stopped at 80 ns and I received a feedback:
>>
>>
>>"ERROR: Calculation halted. Periodic box dimensions have changed too much
>>from their initial values.
>
>What does the density vs time profile for your simulation look like? Have
>the box dimensions indeed changed by a lot? Or, is there any indication of
>an instability (sudden change in energy, etc.)? It is rather unusual to see
>such behavior after 80 ns of simulation, but I'm not a lipid14 user.
>
>....dac
>
>
>_______________________________________________
>AMBER mailing list
>AMBER.ambermd.org
>http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
