Date: Mon, 7 Jun 2021 12:11:34 +0800
Hi everyone,
I'm currently working with protein name 5IBE which has a co-crystallised
ligand 69M and heme cofactor in it. Since the amber force field has no
parameter for non-standard residues and doesn't support my ligand and heme,
I was required to generate parameters using an antechamber tool in amber by
following the tutorial on the amber website:
the tutorial I followed to generate a topology file for my ligand69M:
2.1 Simulating a pharmaceutical compound using Antechamber and the
Generalized Amber Force Field
<http://ambermd.org/tutorials/basic/tutorial4b/index.php>
following this tutorial I successfully generated parameters for my
ligand69M and using the Shahrokh parameter in amber, I was able to generate
topology files for the HEME cofactor in my protein.
But now *my problem is after generating the topology file for both
ligand69M and Heme cofactor, I realised that the numbering in my amino
acids has been shifted for both ligand69M and Heme topology files*
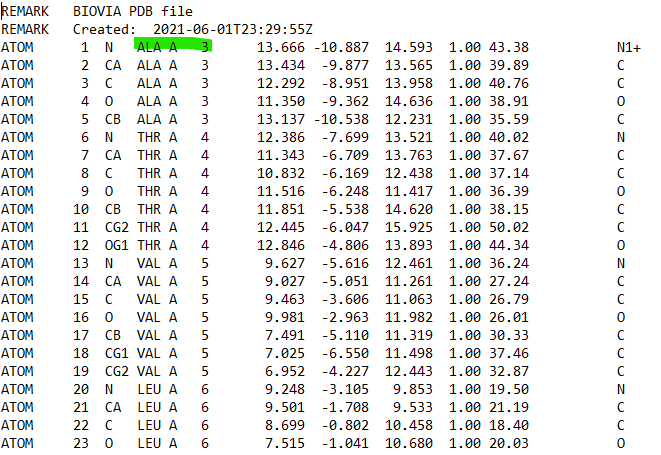
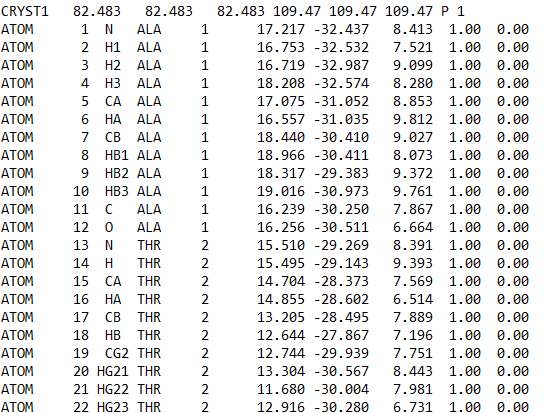
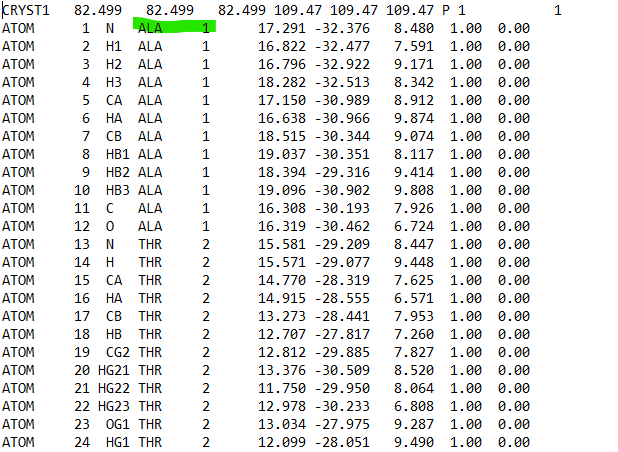
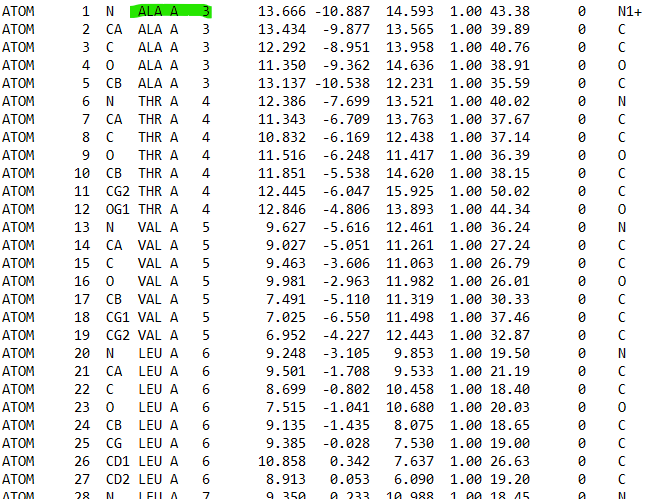
In my initial PDB file, the numbering of amino acid starts with *ALA (first
amino acid) number 3*, but in the topology PDB file created after
generating parameter, the amino acid number was shifted and *ALA starts
with number 1*. So, the whole amino acids numbering sequences in the
topology file has been shifted and not in the original right order. This
has caused the histidine (HIS) and cysteine (CYS ) numbering to also got
shifed in the protein topology file.
p/s: the original PDB that I loaded in tleap had no hydrogen in the
protein. The final topology file created had hydrogen in the protein.
I need to use this topology file for my further work on molecular dynamic
simulation. So, is it a normal phenomenon that can be ignored, or need
attention and have to be fixed before start my further work? Can some
please help me with this, please?
Thank you in advance. Your small guide will help will bring a huge change
to my work.
I have attached the original PDB and topology file of my ligand69M and heme
in the mail. I'm looking for some solution that can solve my problem.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 69m_ori_file_x_hydrogen_.png)
(image/png attachment: 69M_topology.png)
(image/png attachment: heme_topology_file.png)
(image/png attachment: heme_ori_x_h.png)
