Date: Thu, 10 Dec 2020 13:22:30 +0300
Hello amberers,
I am trying to parameterize the B-ob-Si-c3 dihedral. First, I calculated
RESP charges based on HF/6-31G* without ReadRadii and I got "sander
bombed" error during the simulation, hydrogen atoms ran away :) Then I
used ReadRadii (with radius: B 1.99 and Si 2.38) and B3LYP/6-31G* then my
simulation went quite well. But still I have a problem:
1)When I change the charges, even a small amount, the MM scan graph changes
dramatically. But when I tested GAFF, I saw that using mulliken or RESP
almost doesn't affect the chart. So what is wrong with my simulation?
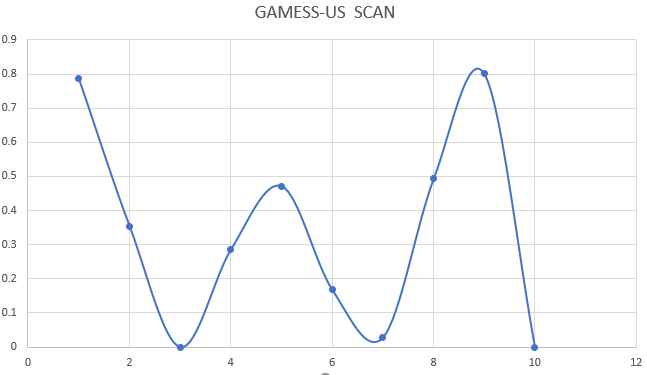
2) the top of the graph is only 0.8kcal / mol and the graph is quite
erratic. Even PAramfit could not find a suitable parameter. So what kind of
way would you suggest me to do this parameterization?
3) Is it OK during parameterization if I use "0" charges for all atoms
instead of using RESP? Or should I rotate dihedral manually and calculate
"single point energy" for each 30 degree step?
Here is my QM scan:
[image: image.png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
