Date: Tue, 1 Dec 2020 13:57:37 +0100
Dear Dr. Case, dear Amber community,
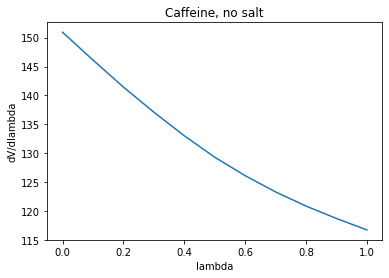
As you expected, the dV/dlambda plot is approximately a straight line
(see attached plot). It's value at lambda=0.5 is 129, which would be
consistent with the resulting free energy assuming a straight dV/dlambda.
However, the molecule does not seem to be charged. Parmed reports:
"Total charge (e-): 0.0000". The pmemd log-file states: "Total charge
of 0.00000199 removed from 24 atoms", and, later:
Sum of charges for TI region 1 = 0.00000199
Skip neutralizing charges...
Sum of charges for TI region 2 = 0.00000000
Skip neutralizing charges...
I tried setting gti_output=1 and gti_cpu_output=0. An example output is:
+++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
Detailed TI info at lambda= 0.50000000000000000
Region H W dH/dl dW/dl
TI 1 vDW -17.92901 0.50000 0.00000 -1.00000
TI 2 vDW -17.92901 0.50000 0.00000 1.00000
lambda = 0.500 : vDW H= -17.9290 dU/dL: L= 0.0000 NL=
0.0000 Tot= 0.00000
------------------------------------------------------------------------
TI 1 Bond 3.60649 0.50000 0.00000 -1.00000
TI 2 Bond 3.60649 0.50000 0.00000 1.00000
lambda = 0.500 : Bond H= 3.6065 dU/dL: L= 0.0000 NL=
0.0000 Tot= 0.00000
------------------------------------------------------------------------
TI 1 Angle 10.47196 0.50000 0.00000 -1.00000
TI 2 Angle 10.47196 0.50000 0.00000 1.00000
lambda = 0.500 : Angle H= 10.4720 dU/dL: L= 0.0000 NL=
0.0000 Tot= 0.00000
------------------------------------------------------------------------
TI 1 Torsion 3.97381 0.50000 0.00000 -1.00000
TI 2 Torsion 3.97381 0.50000 0.00000 1.00000
lambda = 0.500 : Torsion H= 3.9738 dU/dL: L= 0.0000 NL=
0.0000 Tot= 0.00000
------------------------------------------------------------------------
TI 1 EE14-CC -167.78045 0.50000 0.00000 -1.00000
TI 2 EE14-CC 0.00000 0.50000 0.00000 1.00000
lambda = 0.500 : EE14-CC H= -83.8902 dU/dL: L= 167.7805 NL=
0.0000 Tot= 167.78045
------------------------------------------------------------------------
TI 1 VDW14 6.67059 0.50000 0.00000 -1.00000
TI 2 VDW14 6.67059 0.50000 0.00000 1.00000
lambda = 0.500 : VDW14 H= 6.6706 dU/dL: L= 0.0000 NL=
0.0000 Tot= 0.00000
------------------------------------------------------------------------
TI 1 Elec-Rec 70.62218 0.50000 0.00000 -1.00000
TI 2 Elec-Rec 70.71291 0.50000 0.00000 1.00000
lambda = 0.500 : Elec-Rec H= 70.6675 dU/dL: L= 0.0907 NL=
0.0000 Tot= 0.09072
------------------------------------------------------------------------
TI 1 Elec-CC 171.47972 0.50000 0.00000 -1.00000
TI 2 Elec-CC 0.00001 0.50000 0.00000 1.00000
lambda = 0.500 : Elec-CC H= 85.7399 dU/dL: L= -171.4797 NL=
0.0000 Tot= -171.47971
------------------------------------------------------------------------
TI 1 Self-CC 0.00000 0.50000 0.00000 -1.00000
TI 2 Self-CC 137.51100 0.50000 0.00000 1.00000
lambda = 0.500 : Self-CC H= 68.7555 dU/dL: L= 137.5110 NL=
0.0000 Tot= 137.51100
------------------------------------------------------------------------
lambda = 0.500 : Total dU/dl: 133.902458 L: 133.90246 NL: 0.00000
PI: 0.00000
+++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
So it seems the large contributions are Self-CC, Elec-CC and EE14-CC.
Sadly, I am not experienced with TI, so I can't really judge how
"normal" those values are...
Best regards,
Franz Waibl
Am 30.11.2020 um 19:10 schrieb David A Case:
> On Mon, Nov 30, 2020, Franz Waibl wrote:
>> I am trying to compute the free energy of hydration of a set of small
>> molecules at different salt concentrations, using TI.
>>
>> The problem is that I am getting very large free energies (e.g, 130
>> kcal/mol for caffeine, favoring lambda=0).
> Is there any chance that your caffeine molecule has a net charge? It
> would also be helpful to plot <dV/dlambda> vs. lambda: it that
> approximately a straight line? Is that value at lambda = 0.5 about 65?
>
> I'm really guessing here, but this looks like results for de-charging an
> ion. (Use the "summary" command in parmed to look for the charge
> model.)
>
> ....dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: caffeine-electrostatic-ti.png)
