Date: Sat, 31 Oct 2020 14:46:15 +0000
Hello Prof. Case,
I just ran a 100 step minimization to see how the NMR restraints are applied.
So in the min.out file; after the part where the TI masks are read, I found this section on restraints.
Is this getting printed out on the out file because I have set LISTIN=POUT?
I see the 3 distance restraints as mentioned in the file output.disang; but I am still not sure if they are being read properly, why is the NMR restraints bond,angle,torsion all 0 in all the steps of minimization?
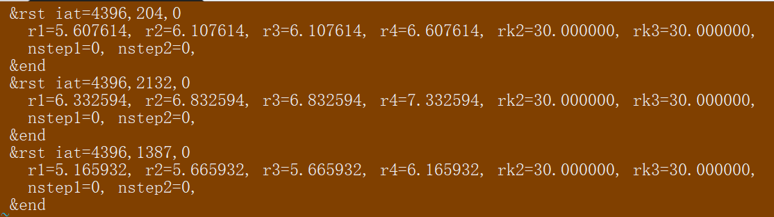
Also this is my output.disang file contents:
[cid:image002.png.01D6AF73.0CEDA8D0]
This is the section of my minimization output:
Ewald parameters:
verbose = 0, ew_type = 0, nbflag = 1, use_pme = 1
vdwmeth = 1, eedmeth = 1, netfrc = 0
Box X = 84.482 Box Y = 81.908 Box Z = 87.672
Alpha = 90.000 Beta = 90.000 Gamma = 90.000
NFFT1 = 90 NFFT2 = 90 NFFT3 = 90
Cutoff= 8.000 Tol =0.100E-04
Ewald Coefficient = 0.34864
Interpolation order = 4
TI Mask 1 :IPA; matches 12 atoms
TI Mask 2 matches 0 atoms
TI region 1: 50200 atoms
TI region 2: 50188 atoms
SC Mask 1 :IPA; matches 12 atoms
--------------------------------------------------------------------------------
3. ATOMIC COORDINATES AND VELOCITIES
--------------------------------------------------------------------------------
default_name
begin time read from input coords = 0.000 ps
Begin reading energy term weight changes/NMR restraints
WEIGHT CHANGES:
DUMPFREQ 1 0 0.000000 0.000000 0 0
** No weight changes given **
RESTRAINTS:
Requested file redirections:
DISANG = output.disang
DUMPAVE = dist_0.00922.dat
LISTIN = POUT
LISTOUT = POUT
Restraints will be read from file: output.disang
Here are comments from the DISANG input file:
******
C2 ( 4396)-CA ( 204) NSTEP1= 0 NSTEP2= 0
R1 = 5.608 R2 = 6.108 R3 = 6.108 R4 = 6.608 RK2 = 30.000 RK3 = 30.000
Rcurr: 6.313 Rcurr-(R2+R3)/2: 0.205 MIN(Rcurr-R2,Rcurr-R3): 0.205
******
C2 ( 4396)-CA ( 2132) NSTEP1= 0 NSTEP2= 0
R1 = 6.333 R2 = 6.833 R3 = 6.833 R4 = 7.333 RK2 = 30.000 RK3 = 30.000
Rcurr: 6.883 Rcurr-(R2+R3)/2: 0.050 MIN(Rcurr-R2,Rcurr-R3): 0.050
******
C2 ( 4396)-CA ( 1387) NSTEP1= 0 NSTEP2= 0
R1 = 5.166 R2 = 5.666 R3 = 5.666 R4 = 6.166 RK2 = 30.000 RK3 = 30.000
Rcurr: 5.641 Rcurr-(R2+R3)/2: 0.025 MIN(Rcurr-R2,Rcurr-R3): 0.025
Number of restraints read = 3
Done reading weight changes/NMR restraints
Number of triangulated 3-point waters found: 15264
Number of shake restraints removed in TI region 1 : 0
Number of shake restraints removed in TI region 2 : 0
*********************************************************************************************************************************************************************
From: David A Case<mailto:david.case.rutgers.edu>
Sent: 30 October 2020 21:22
To: AMBER Mailing List<mailto:amber.ambermd.org>
Subject: Re: [AMBER] NMR restraints
On Fri, Oct 30, 2020, Debarati DasGupta wrote:
>I was trying to set up a minimization (TI run) and I see that NMR bond
>angle Torsion energies are all 0.
>LISTIN=POUT
To add to what Carlos said: if you set LISTIN=POUT, you should see a
detailed printout of all the NMR calculations on the initial step. You
can check to see if they are defined the way you want, and see how the
energies are computed.
[If for some reason this info doesn't show up in parallel, just run a
single step calculation in serial mode.]
....dac
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: FE11AF737B1C4870B092E7D9DCFB9142.png)
