Date: Mon, 26 Oct 2020 08:21:15 +0000
Monday morning started with a bit of celebration. I did not have access to GUI evaluation over the weekend so I based my judgement regarding the failed minimisation on the output of the perl scripts.
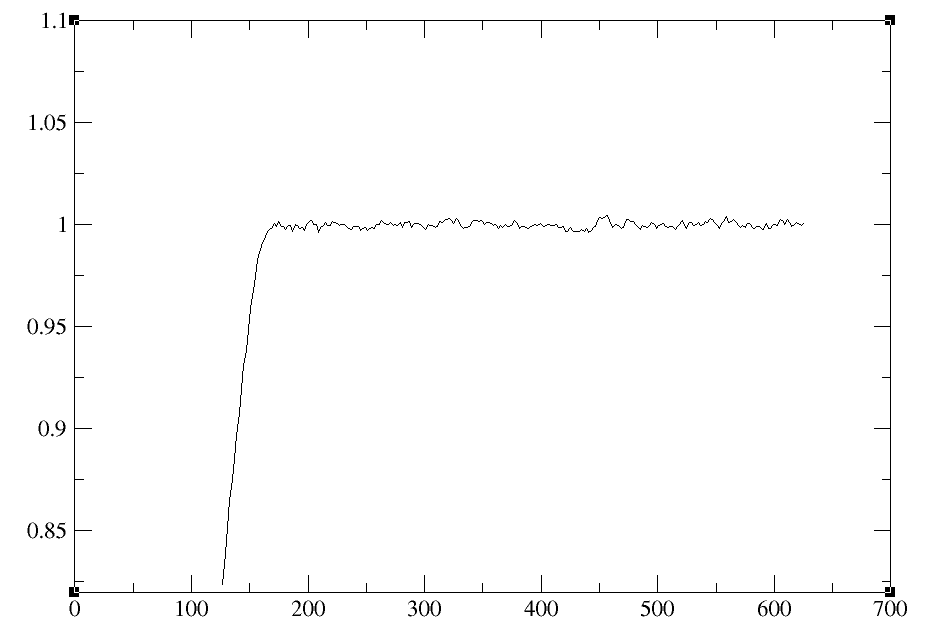
Looking at the system with VMD shows water and peptide molecules moving around as expected and a box that equilibrates nicely even prior to barostat=2 and a short taup. It seems that the order of sourcing in leap really was the issue and switching the order solved the problem.
Apparently, the perl script is not very forgiving so the CMAP apparently effects some indexing. I have not worked with perl at all so I have no idea where the problem lies though at some point in time when I have time I will try to fix this.
Meanwhile, the usual horrible oneliner solved the problem intermediately:
paste <(grep "TIME(PS)" eqPres.out | awk '{print $6}') <(grep "Density" eqPres.out | awk '{print $3}') | column -s $'\t' -t | head -n $(( $(grep "TIME(PS)" eqPres.out | awk '{print $6}' | wc -l)-2 )) >> summary.DENSITY
[cid:9F5AB618-40CC-4EF1-AB97-42B5EC652952.client.lnu.se]
Thank you both for all you help, feedback and input.
Best regards
// Gustaf
On 24 Oct 2020, at 21:58, Carlos Simmerling <carlos.simmerling.gmail.com<mailto:carlos.simmerling.gmail.com>> wrote:
is the problem now just in extracting things from the mdout file?
use of ff19SB does add a "CMAP" term to the energy output. so anything that
assumes all of the energy terms are in specific places might not work.
I imagine if you look at the file and compare to the other one you might be
able to adjust the script, or use a combination of grep and awk to extract
what you want. I don't use that perl script so can't really comment on how
flexible it is.
here is an example from a system where did MD with both force fields:
ff14SB:
NSTEP = 1000 TIME(PS) = 2101.000 TEMP(K) = 298.75 PRESS =
15.9
Etot = -44184.4117 EKtot = 10921.8008 EPtot =
-55106.2125
BOND = 442.7755 ANGLE = 1161.2657 DIHED =
1903.5578
1-4 NB = 552.1177 1-4 EEL = 4678.8785 VDWAALS =
5871.5787
EELEC = -69716.3864 EHBOND = 0.0000 RESTRAINT =
0.0000
EKCMT = 4641.7222 VIRIAL = 4581.2633 VOLUME =
175735.8591
Density =
1.0355
------------------------------------------------------------------------------
ff19SB:
NSTEP = 1000 TIME(PS) = 3001.000 TEMP(K) = 296.65 PRESS =
58.2
Etot = -59114.2876 EKtot = 10887.3496 EPtot =
-70001.6372
BOND = 439.2101 ANGLE = 1197.8938 DIHED =
908.4324
UB = 0.0000 IMP = 0.0000 CMAP =
135.9763
1-4 NB = 560.3848 1-4 EEL = 4715.4626 VDWAALS =
7598.8540
EELEC = -85557.8511 EHBOND = 0.0000 RESTRAINT =
0.0000
EKCMT = 4645.2896 VIRIAL = 4425.1982 VOLUME =
175073.8963
Density =
1.0436
------------------------------------------------------------------------------
On Sat, Oct 24, 2020 at 3:49 PM Gustaf Olsson <gustaf.olsson.lnu.se<mailto:gustaf.olsson.lnu.se>> wrote:
So, I corrected the priority order, rebuilt and tried.
Nothing has finished yet though as I am looking at a completely blank
output from energy minimization with "process_m*.perl" I think there will
be no difference from before.
The only thing left that I can think of is the amber version. Obviously, I
can optimize my input in more than one way and try different combinations
of barostat and taup settings though if nothing is moving at all, even at
40 bar and a taup of 0.1, then I don't think this tweaking the input
settings will change much.
I also checked and I am up to date on patches and updates for amber18.
So, the OPC water model works just fine. However, the combination with
ff19SB breaks something. Both of you managed to run the simulations, are
you using amber20? If so, I suppose I will have to start asking for a
license to be purchased and potentially this will solve my problems(?).
If either of you could be so kind as to look in the newly added "4
desperation/ff19SB_opc" folder and just try to run the content of
"cloudJobEQ.sh" "as is", preferably as CPU run, I would be extremely
grateful. To really push the boundaries here, if you manage to run the
provided files "as is", could you also run the "process_m*.perl" scripts
and see of you get the expected output?
You should both have read/write access to the resources so you can
download/upload any files there and provide notes/feedback.
Thank you in advance and have a fantastic rest of your weekend.
Best regards
// Gustaf
________________________________
Från: Carlos Simmerling <carlos.simmerling.gmail.com<mailto:carlos.simmerling.gmail.com>>
Skickat: den 23 oktober 2020 19:28:58
Till: AMBER Mailing List
Ämne: Re: [AMBER] ff19SB and OPC, process_(min/md)out.perl and LEaP
to follow up on Dave's suggestion - I tried Gustaf's low density input
coordinate with barostat=1 + taup=0.1, and also barostat=2.
Barostat=2 gave about 5-10x increase in the slope of density vs time.
carlos
On Thu, Oct 22, 2020 at 8:48 AM David A Case <david.case.rutgers.edu<mailto:david.case.rutgers.edu>>
wrote:
On Wed, Oct 21, 2020, Carlos Simmerling wrote:
what is your pressure coupling constant? I often get vacuum bubbles
unless
I include a step in my equilibration with tight coupling, taup around
0.1
or smaller.
Carlos: have you ever tried barostat=2? It would be interesting to know
if that makes a difference. Also, what is your starting density? If
you use a closeness parameter of 0.7--0.8 (in solvateBox or solvateOct),
you'll get a higher starting density, which might(?) reduce the tendency
to get bubbles.
....dac
_______________________________________________
AMBER mailing list
AMBER.ambermd.org<mailto:AMBER.ambermd.org>
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org<mailto:AMBER.ambermd.org>
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org<mailto:AMBER.ambermd.org>
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_2020-10-26_at_09.19.50.png)
