Date: Thu, 15 Oct 2020 10:11:28 -0700
Dear Callum sir,
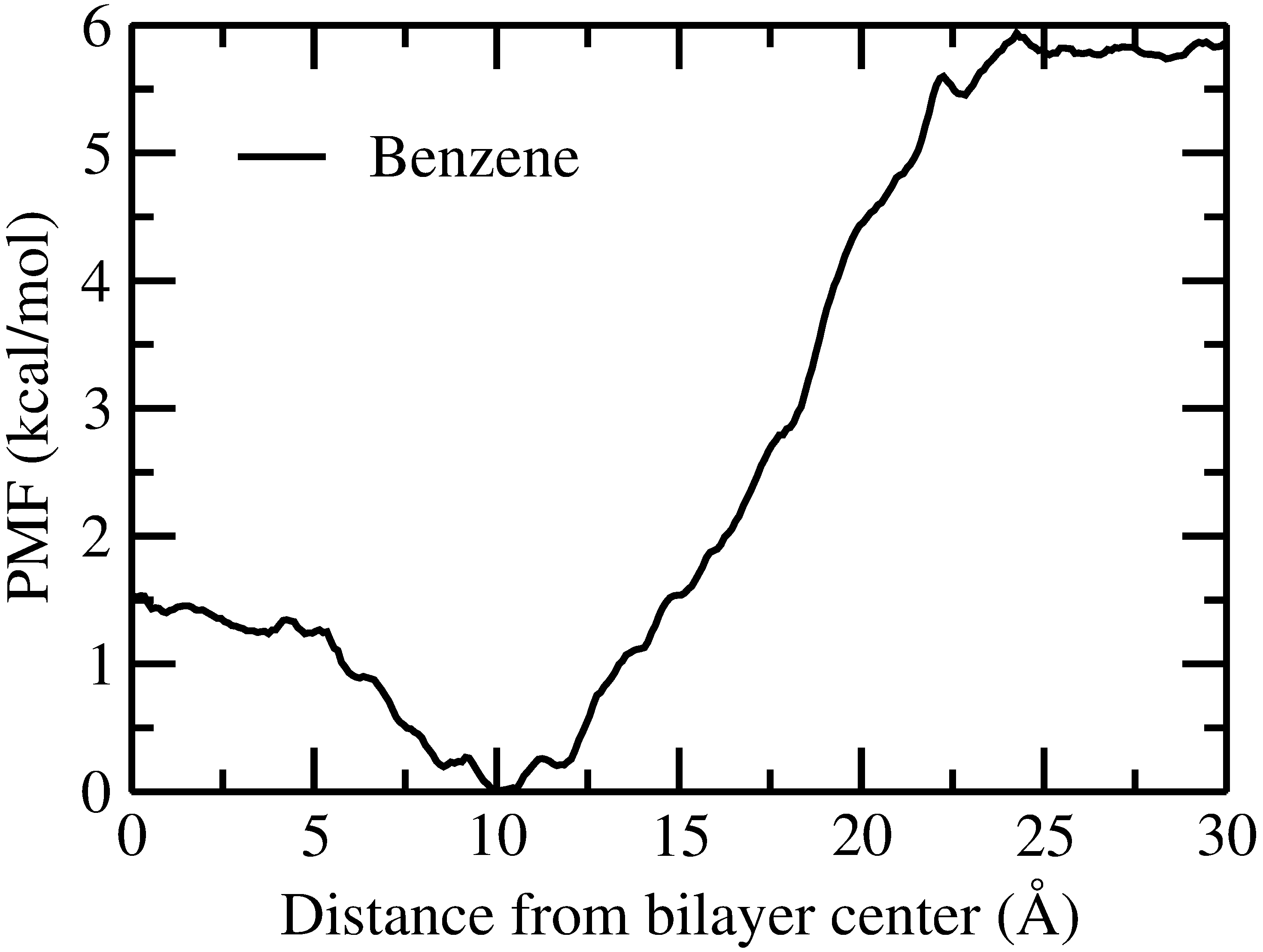
Hope you are doing well. As you suggested I tried with benzene and got the
following graph.
This time also I didn't get any negative value. I also attached input files
and RST files for your reference.
Kindly take a look and please let me know if I did something wrong. Thank
you.
Regards
Somdev Pahari
On Tue, Sep 29, 2020 at 11:20 AM somdev pahari <somdevpahari1.gmail.com>
wrote:
> Thank you Callum sir for your quick response and detailed explanation.
> I forgot that there were two different force fields in the tutorial and in
> the publication, that's my bad.
> I will definitely try with benzene and will let you know if anything finds
> unusual.
>
> Regards
> Somdev Pahari
>
> On Mon, Sep 28, 2020 at 1:38 PM Dickson, Callum <
> callum.dickson.novartis.com> wrote:
>
>> Hi Somdev,
>>
>> Your results look quite reasonable but you will need to simulate longer
>> to get convergence. Why do you think you should have negative values?
>> Debasing the results so that dG=0 in the water phase, a negative region
>> would indicate methanol prefers the lipid bilayer environment over water.
>>
>> If you look at the PMF curve in the tutorial, although it comes back down
>> close to zero, at no point is the free energy negative, indicating methanol
>> would prefer to remain in water using GAFF and lipid14 force fields. In the
>> Orsi paper, it seems like the free energy for methanol is very slightly
>> negative around the lipid head group region, however these simulations used
>> a coarse grained membrane so it is hard to directly compare.
>>
>> The logP for methanol is around logP = -0.5, so it prefers water over
>> oil. logP doesn't always track directly with membrane partitioning but it
>> is a good indicator. If you run the same PMF for something like benzene
>> (logP ~ 2), it should certainly prefer the membrane environment with -ve
>> free energy.
>>
>> Best,
>> Callum
>>
>> -----Original Message-----
>> From: somdev pahari <somdevpahari1.gmail.com>
>> Sent: Tuesday, September 29, 2020 4:25 AM
>> To: AMBER Mailing List <amber.ambermd.org>
>> Subject: [AMBER] PMF calculation
>>
>> Dear Amber users,
>> I am trying to calculate PMF for the lipid membrane system and using this
>> tutorial as a reference (
>> https://urldefense.proofpoint.com/v2/url?u=https-3A__github.com_callumjd_AMBER-2DUmbrella-5FCOM-5Frestraint-5Ftutorial&d=DwIBaQ&c=ZbgFmJjg4pdtrnL2HUJUDw&r=HwsFjSfOtLupDR-NuCP430rdz1DD2LkJxNM3BsKSjrw&m=glKztQJkVaVEw8x6mt5z6eAYHOsRFnnr0EC8hgMFGK8&s=xtdgAIY30mJZ3ga2YeZkK2GDHVS9g672O7YCsr3B8xs&e=
>> ) with Amber 18.
>> I am trying to reproduce some results as described in the tutorial as
>> well as in this paper (
>> https://urldefense.proofpoint.com/v2/url?u=https-3A__doi.org_10.1021_jp903248s&d=DwIBaQ&c=ZbgFmJjg4pdtrnL2HUJUDw&r=HwsFjSfOtLupDR-NuCP430rdz1DD2LkJxNM3BsKSjrw&m=glKztQJkVaVEw8x6mt5z6eAYHOsRFnnr0EC8hgMFGK8&s=QLU7ZMNOXG81ve1WJAFSk8mN9-x4TIz5JiQYCmMPC14&e=
>> ), also mentioned in the tutorial.
>> I did it for water and methanol. What I faced that in the case of
>> methanol there do not have any single negative value as it should be. I
>> took 0.5 A as window width and ran a test simulation of 5 ns for each. I
>> know that it's not enough for convergence, still, am I missing something?
>> Kindly help me out.
>> Thanks in advance.
>>
>> Regards
>> Somdev Pahari
>> Research Scholar
>> NIT Rourkela
>> Odisha, India
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: COM_dist.RST
- application/octet-stream attachment: COM_pull.RST
- application/octet-stream attachment: 05_Pull.in
- application/octet-stream attachment: 06_Prod.in
(image/jpeg attachment: benzene.jpg)
