Date: Sun, 4 Oct 2020 13:55:41 +0000
Hello Dan,
But here is the crux of the issue I am facing ( rather issues!)..
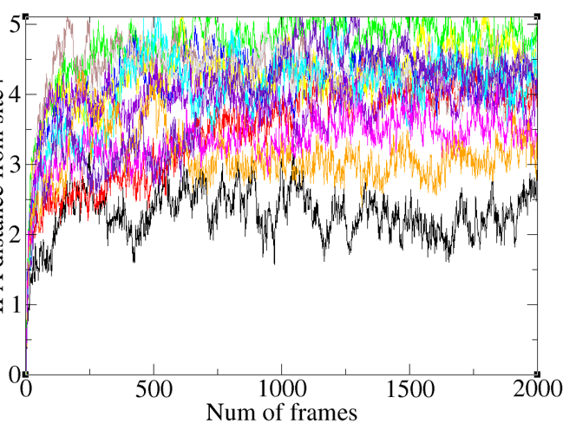
While applying a restraint of 10kcal/mol on my C3N ligand, why does the rms fluctuations look like this...( Plot of 12 lambdas C3N RMSD over the production phase of TI runs)??? Any ideas you have as to why I am doing wrong? Sorry I don’t have axis labels, but I am totally lost as to why my ligand meanders so much inspite of having a rock-solid (ntr =1, restraintmask=’2,23,271,274’) 274 is my C3N and the others are protein residues...
Any help or suggestions on how to fix this weird behaviour?
Debarati
[cid:image003.png.01D69A34.82D96330]
Sent from Mail<https://go.microsoft.com/fwlink/?LinkId=550986> for Windows 10
From: Daniel Roe<mailto:daniel.r.roe.gmail.com>
Sent: 04 October 2020 09:48
To: AMBER Mailing List<mailto:amber.ambermd.org>
Subject: Re: [AMBER] RMSD of ligand in pocket
Hi,
You've got 2 rms commands, the first one fits on residues 2 to 272 of
your protein, the next is the no-fit rmsd of your ligand (i.e. the
RMSD of the ligand in the reference frame of the protein), which seems
like what you want. Does this answer your question?
-Dan
On Sat, Oct 3, 2020 at 11:08 AM Debarati DasGupta
<debarati_dasgupta.hotmail.com> wrote:
>
> Hello All,
>
> I have a kinase and a acetonitrile bound to the surface of the protein.
>
> I am trying to analyse the movement of the ligand from the pocket during my TI runs, for each lambda window.
>
> Here is a sample input file I am using for lambda=0.00922
>
>
>
> parm C3N_solvated.prmtop
>
> trajin C3N_solvated_prod.mdcrd 1 last 1
>
> center mass origin :1-273
>
> image origin center
>
> reference ../complex_WAT_box_SITE1.pdb
>
> strip :WAT
>
> strip :Na+
>
> rms reference mass out backbone_0.00922_aligned_RMSD.txt :2-272.CA,C,N
>
> rms C3N_site1 :C3N&!.H= nofit mass out lig_prob_0.00922_rmsd.dat
>
> go
>
>
>
> Residue 1 and 273 are my ACE and NME caps respectively...Residue 2 to 272 is my protein atoms.
>
> I am not sure if I should use “nofit” in my rms command line?
>
> Any insights would be super helpful.
>
> Thanks
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: F5698954F188421F96A548D21CEB98A2.png)
