Date: Sat, 9 May 2020 11:56:04 +0000
Hello everyone,
Is there anyone here on the mailing list who has done extensive absolute free energy TI calculations; specifically :NPT equilibrations on GPUs and have seen success?
This is in reference to Thermodynamic integration calculations on AMBER18.
Well, I have been working on it last ~5-6 months and it seems and whatever I try, the 2ns of density equilibration (ntp=1) ALWAYS fails on the GPUs.
That has become a real issue as its taking 30-36 hours on 16 cpu processors to perform 2ns of NPT equilibration. That is really making the project unfeasible. I have 2 kinase proteins and 12 lambdas each and there are 4-5 different sites on each protein that I am interested in computing absolute binding free energies for...
I see there is a paper in ACS Omega (published ~March 2020) which does GPU-TI but I m not sure as the protocol is not clear.
As for my cases... the minimization, gradual heating, everything happens nicely on GPUs, but the moment I switch to ntp=1, the error I always get is
“INVALID MEMORY ACCESS ENCOUNTERED..SEG FAULT”....
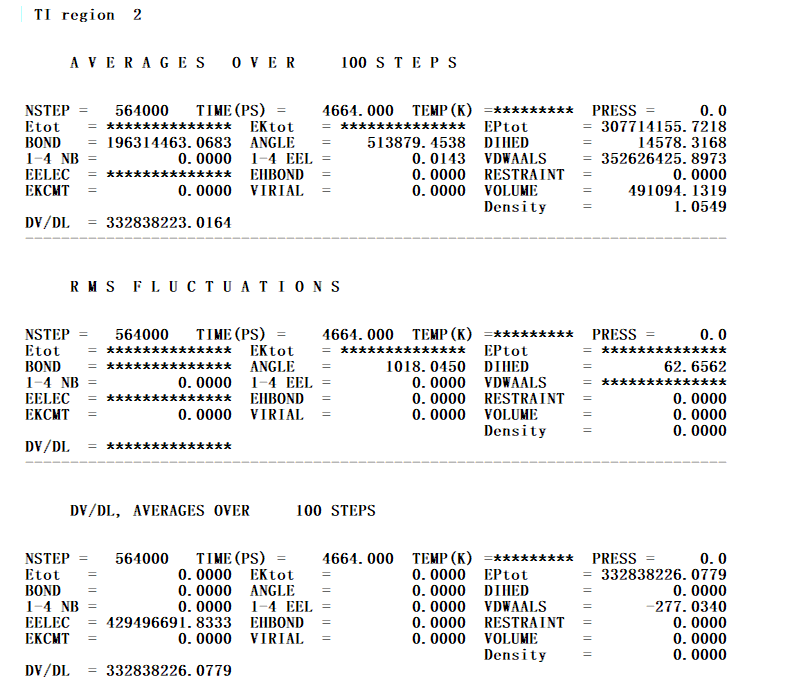
The energies all become *********, DVDL=***** everything just crashes. A small snapshot of the values attached:
[cid:image001.png.01D625D7.27533850]
Any slight suggestions/ideas/troubleshooting will help immensely.
Thank you all.
Regards
Debarati
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 58F0691D06424AE4BEAB224E4B20E7C9.png)
