Date: Mon, 4 May 2020 09:13:53 -0400
Hello,
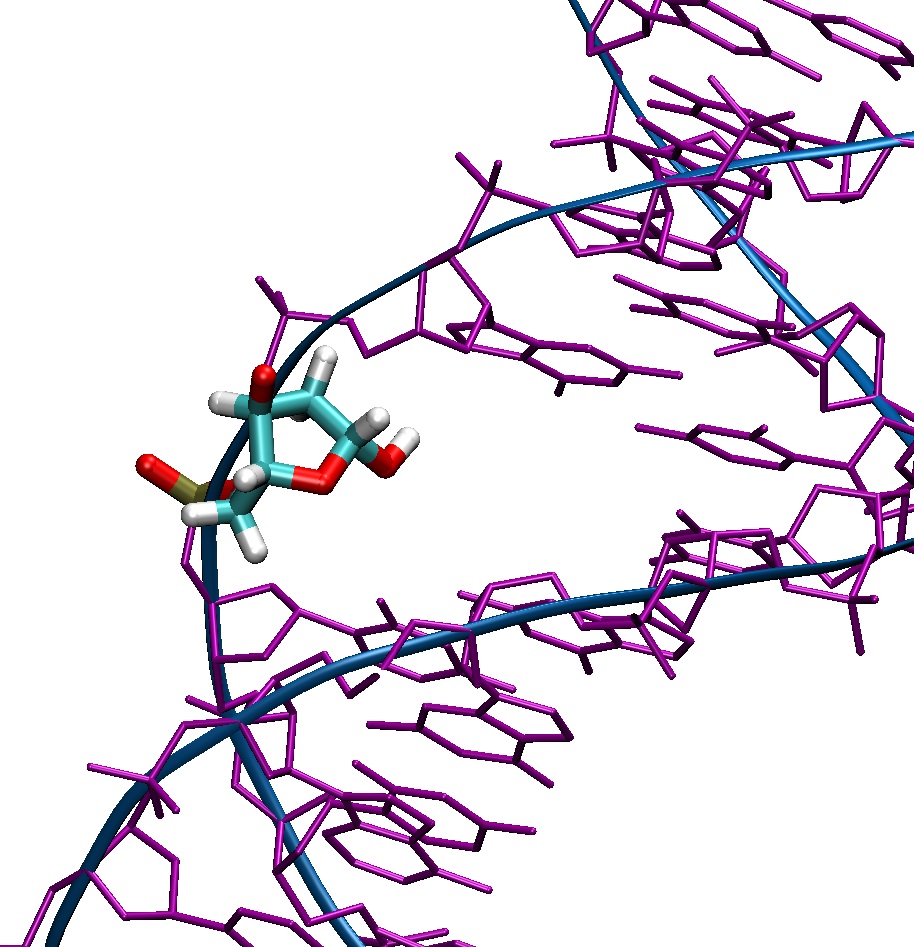
The hydroxyl at C1' is a result of removal of the base after oxidative
damage.
In reality, the abasic/apurinic site exists as an equilibrium between a
furanose ring with a hydroxyl group, and an opened ring alcohol and
aldehyde form.
The library files provided return the correct abasic site chemistry for the
furanose ring form (Chen et al., Biochemistry 2007, 46, 3096-3107, Scheme
1, "normal abasic site").
If you want the open ring structure, you'll probably need to parameterize
it as a special residue yourself, or do a literature search for that moiety
- I do not know of any library files for this.
Otherwise, the hydroxyl group should be there and I'm not sure what you are
considering an error.
-Christina
On Mon, May 4, 2020 at 8:40 AM Robert Molt <rwmolt07.gmail.com> wrote:
> Good evening,
>
> I am observing a problem of added atoms I do not desire after leap
> processing; I am hoping for assistance in tracking down the mistake I am
> making which results in these extra atoms.
>
> Consider a protein/DNA complex given in the attached file 5.pdb. It has
> standard residues, but for the "ABB residue." Dr. Bergonzo kindly offered
> me a library file with parameters for an ABB “residue” (I re-post her file
> here as ABB.off). I loaded the initial pdb structure, with the leap
> commands specified below (I have also attached the leap.log file). I had no
> particular errors within leap (beyond what I believe are “innocent”
> warnings in the leap.log). I then proceeded to minimize and heat the
> system, and finally observed a problem: leap added a OH in the C1 position
> of the ABB residue. This was not intended. The file “stripped.pdb” shows
> the system post-heating (post-leap, moreover).
>
> I believe the leap log clarifies the point of departure:
>
> "Joining DC - ABB
> Added missing heavy atom: .R<ABB 329>.A<O1B 13>
> Joining ABB - DC"
>
> Questions:
>
> 1.) I believe that this line from the leap log shows the adding the OH
> group I do not wish. Can this interpretation be confirmed?
>
> 2.) This would imply to me that my ABB.off file is expecting a OH residue
> on the C1 atom. When I examine the .off file, I see an O1B specified in the
> line
>
> "O1B" "OH" 0 1 131072 13 8 -0.648200
>
> Is this interpretation correct? I have been trying to find the part of the
> AMBER manual which clarifies this file format, but am coming up short. I
> would think that this could be found in chapter 15, beginning page 225 of
> the 19th edition, but I do not see anything clarifying the .off file
> format. I also cannot find anything in the manual when I search for .off
>
> 3.) I am very confused how the attached .off file would result in an O1B
> being added to a C1’ carbon. Perhaps this answer will be forthcoming in
> better understanding the file format of .off files.
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- ----------------------------------------------------------------- Christina Bergonzo Research Chemist Biomolecular Measurement Division, MML, NIST -----------------------------------------------------------------
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: abasic-site.jpg)
