Date: Mon, 17 Feb 2020 11:25:51 +0900
Dear All,
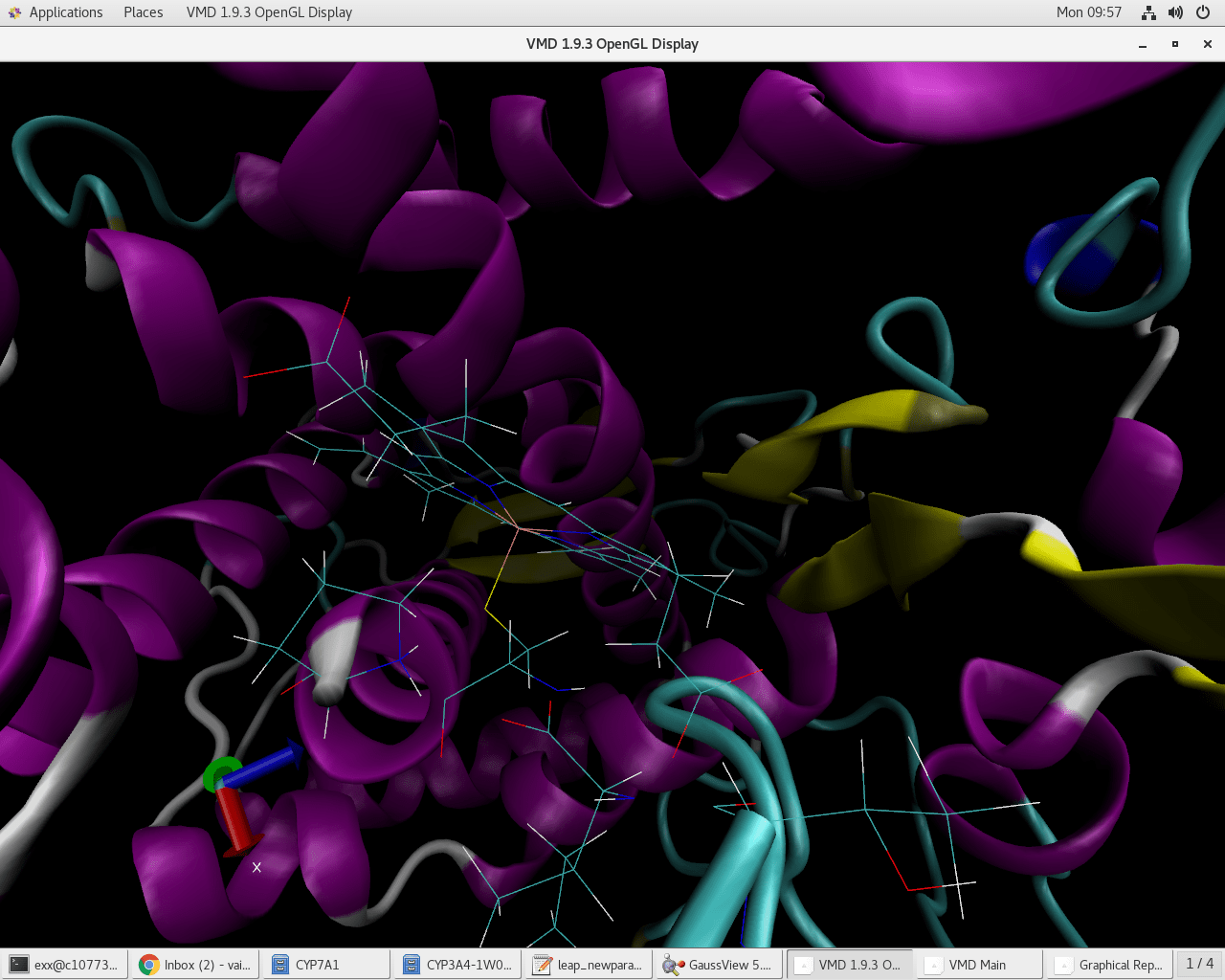
The vmd shows the lack of bonds upon loading prmtop and rst7 files
(attached compressed image).
Thus the expected connection (bond) was not created even after giving
correct atom-residue information as shared in earlier email.
Can someone please suggest on how to fix this problem?
Also I'm getting errors while writing pdb using ambpdb with -conect option.
thank you.
ambpdb -p test.prmtop -c test.rst7 -conect > 3v8d-test8.pdb
Error: Atom 7779 was assigned a lower molecule # (1) than previous atom (2).
Error: This can happen if bond information is incorrect or missing, or if
the
Error: atom numbering in molecules is not sequential. Try one of the
Error: following:
Error: - If this is a PDB file, try using the 'noconect' keyword.
Error: - If this topology did not have bond info, try increasing the bond
Error: search cutoff above 0.2 Ang. ('bondsearch <cutoff>').
Error: - Use the 'fixatomorder' command to reorder the topology and any
Error: associated coordinates.
Error: - Use the 'setMolecules' command in parmed to reorder only the
Error: topology.
Error: Could not determine molecule information for test.prmtop.
Error: SetSolventInfo: No molecule information.
Error: Could not determine solvent information for test.prmtop.
On Mon, Feb 17, 2020 at 3:55 AM Busra Demir <bdemir09.gmail.com> wrote:
> Hi Vaibhav,
>
> This is probably about VMD itself. It has cutoff distance to describe a
> bond.
> You should load both .prmtop and .inpcrd files to be sure of leap bonding.
>
> Best,
>
> Busra Demir
> Bionanodesign Laboratory, TOBB ETU
> Sogutozu/Ankara
>
>
> On Sun, Feb 16, 2020 at 7:28 PM Vaibhav Dixit <vaibhavadixit.gmail.com>
> wrote:
>
> > Dear Prof. Carlos SImerling and other experts,
> > I build the non-standard residue (Cys-Heme) by running G09 calculations
> and
> > esp file. The Cys was capped with N*Me* and CO-*H*.
> > Then I used antechamber to generate mol2 file and used -a gaff -nc -2 -m
> 6
> > options.
> > Then I edited the mol2 in xleap to delete capping atoms and kept only the
> > NHCH(CH2S)CO of the Cys and bonded the S and porphyrin Ns with Fe. This
> > gave me uncapped Cys-Heme mol2 using which I ran prmchk2 to generate
> frcmod
> > and added remaining missing parameters from SI of this paper
> > <https://www.ncbi.nlm.nih.gov/pubmed/21997754>. That is why I was
> looking
> > for CYS and CYX parameters in the last 2-3 days.
> >
> > Other people have parameterized heme and bonded the S to Fe, but I want
> to
> > parameterize the whole CYS-Heme and connect it with the rest of the
> > backbone using CYS's N and C atoms.
> > Please do give your valuable inputs on if this approach is reasonably
> > correct or not.
> > Thank you very much
> >
> > On Sun, Feb 16, 2020 at 8:59 PM Carlos Simmerling <
> > carlos.simmerling.gmail.com> wrote:
> >
> > > You don't say how you built the non standard residue. The leap output
> > > doesn't really tell us your while workflow. If you used gaff, you'll
> need
> > > cross terms for the bonded parameters between gaff and ff14SB, or
> > something
> > > else depending on what you used.
> > > Also in Vmd make sure you are not evaluating bonds based on the pdb.
> You
> > > must load the prmtop and coordinates from leap in order to display the
> > > bonds present in the prmtop file.
> > >
> > > On Sun, Feb 16, 2020, 10:01 AM Vaibhav Dixit <vaibhavadixit.gmail.com>
> > > wrote:
> > >
> > > > Dear All,
> > > > I'm using the attached pdb, mol2 and frcmod files in tleap and giving
> > the
> > > > following commands, but I can't see expected bonds in vmd.
> > > > I have double checked that my usage in bond command is correct (at
> > least
> > > to
> > > > me).
> > > > I get 1 non-fatal error and some warnings but when I save pdb the
> > > terminal
> > > > residues don't appear to have bonded as then confirmed visually in
> vmd.
> > > > Can you please suggest, why these bond commands are not working as
> > > > expected?
> > > > thank you.
> > > >
> > > > Here are my tleap commands.
> > > > source leaprc.protein.ff14SB
> > > > source leaprc.gaff
> > > > loadamberparams frcmod.ionsjc_tip3p
> > > > loadamberparams test-uncap1.frcmod
> > > > HEM = loadmol2 test-uncap1.mol2
> > > > 3v8d = loadpdb 3v8d-test7.pdb
> > > >
> > > >
> > > >
> > > > Warning: Close contact of 1.370191 angstroms between .R<GLU
> 447>.A<HB2
> > 6>
> > > > and .R<ALA 450>.A<HA 4>
> > > > Checking parameters for unit '3v8d'.
> > > > Checking for bond parameters.
> > > > Checking for angle parameters.
> > > > check: Errors: 1 Warnings: 25
> > > >
> > > > *> bond 3v8d.482.C3 3v8d.421.N> bond 3v8d.420.C 3v8d.482.N1*
> > > > > saveamberparm 3v8d test.prmtop test.rst7
> > > > Checking Unit.
> > > >
> > > > Warning: There is a bond of 4.825647 angstroms between:
> > > >
> > > > Warning: The unperturbed charge of the unit (3.077800) is not
> integral.
> > > >
> > > > Warning: The unperturbed charge of the unit (3.077800) is not zero.
> > > >
> > > > Note: Ignoring the error and warnings from Unit Checking.
> > > >
> > > > Building topology.
> > > > Building atom parameters.
> > > > Building bond parameters.
> > > > Building angle parameters.
> > > > Building proper torsion parameters.
> > > > Building improper torsion parameters.
> > > > total 1612 improper torsions applied
> > > > Building H-Bond parameters.
> > > > Incorporating Non-Bonded adjustments.
> > > > Not Marking per-residue atom chain types.
> > > > Marking per-residue atom chain types.
> > > > (Residues lacking connect0/connect1 -
> > > > these don't have chain types marked:
> > > >
> > > > res total affected
> > > >
> > > > CHIE 1
> > > > CILE 1
> > > > HEM 1
> > > > NPRO 1
> > > > NSER 1
> > > > )
> > > > (no restraints)
> > > > > savepdb 3v8d 3v8d-tleap.pdb
> > > > Writing pdb file: 3v8d-tleap.pdb
> > > >
> > > > Warning: Converting N-terminal residue name to PDB format: NSER ->
> SER
> > > >
> > > > Warning: Converting C-terminal residue name to PDB format: CILE ->
> ILE
> > > >
> > > > Warning: Converting N-terminal residue name to PDB format: NPRO ->
> PRO
> > > >
> > > > Warning: Converting C-terminal residue name to PDB format: CHIE ->
> HIE
> > > > > quit
> > > > Quit
> > > >
> > > > --
> > > >
> > > > Regards,
> > > >
> > > > Dr. Vaibhav A. Dixit,
> > > >
> > > > Visiting Scientist at the Manchester Institute of Biotechnology
> (MIB),
> > > The
> > > > University of Manchester, 131 Princess Street, Manchester M1 7DN, UK.
> > > > AND
> > > > Assistant Professor,
> > > > Department of Pharmacy,
> > > > ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄
> > > > Birla Institute of Technology and Sciences Pilani (BITS-Pilani),
> > > > VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031.
> > > > India.
> > > > Phone No. +91 1596 255652, Mob. No. +91-7709129400,
> > > > Email: vaibhav.dixit.pilani.bits-pilani.ac.in,
> vaibhavadixit.gmail.com
> > > > http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile
> > > > https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/
> > > >
> > > > ORCID ID: https://orcid.org/0000-0003-4015-2941
> > > >
> > > > http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra
> > > >
> > > > P Please consider the environment before printing this e-mail
> > > > _______________________________________________
> > > > AMBER mailing list
> > > > AMBER.ambermd.org
> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > >
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> >
> >
> > --
> >
> > Regards,
> >
> > Dr. Vaibhav A. Dixit,
> >
> > Visiting Scientist at the Manchester Institute of Biotechnology (MIB),
> The
> > University of Manchester, 131 Princess Street, Manchester M1 7DN, UK.
> > AND
> > Assistant Professor,
> > Department of Pharmacy,
> > ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄
> > Birla Institute of Technology and Sciences Pilani (BITS-Pilani),
> > VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031.
> > India.
> > Phone No. +91 1596 255652, Mob. No. +91-7709129400,
> > Email: vaibhav.dixit.pilani.bits-pilani.ac.in, vaibhavadixit.gmail.com
> > http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile
> > https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/
> >
> > ORCID ID: https://orcid.org/0000-0003-4015-2941
> >
> > http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra
> >
> > P Please consider the environment before printing this e-mail
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
>
>
> --
> Büşra
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Regards, Dr. Vaibhav A. Dixit, Visiting Scientist at the Manchester Institute of Biotechnology (MIB), The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. AND Assistant Professor, Department of Pharmacy, ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄ Birla Institute of Technology and Sciences Pilani (BITS-Pilani), VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031. India. Phone No. +91 1596 255652, Mob. No. +91-7709129400, Email: vaibhav.dixit.pilani.bits-pilani.ac.in, vaibhavadixit.gmail.com http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/ ORCID ID: https://orcid.org/0000-0003-4015-2941 http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra P Please consider the environment before printing this e-mail
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image-min.png)
