From: Aravind R <aravindspg27.gmail.com>
Date: Mon, 7 Oct 2019 12:57:12 +0530
Dear Amber users,
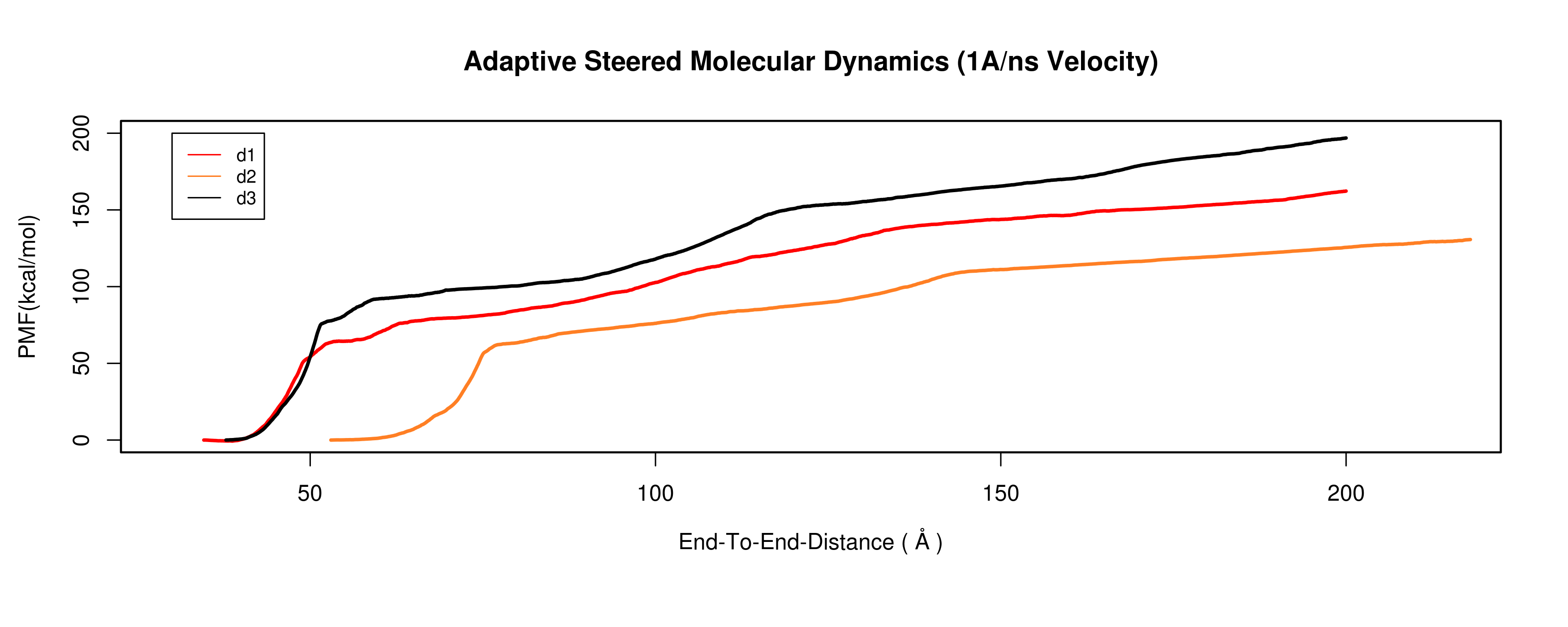
I performed ASMD simulation on three proteins to completely unfold them
from a fully folded conformation.
I followed the ASMD tutorial, and I constructed PMF from work done (fig1).
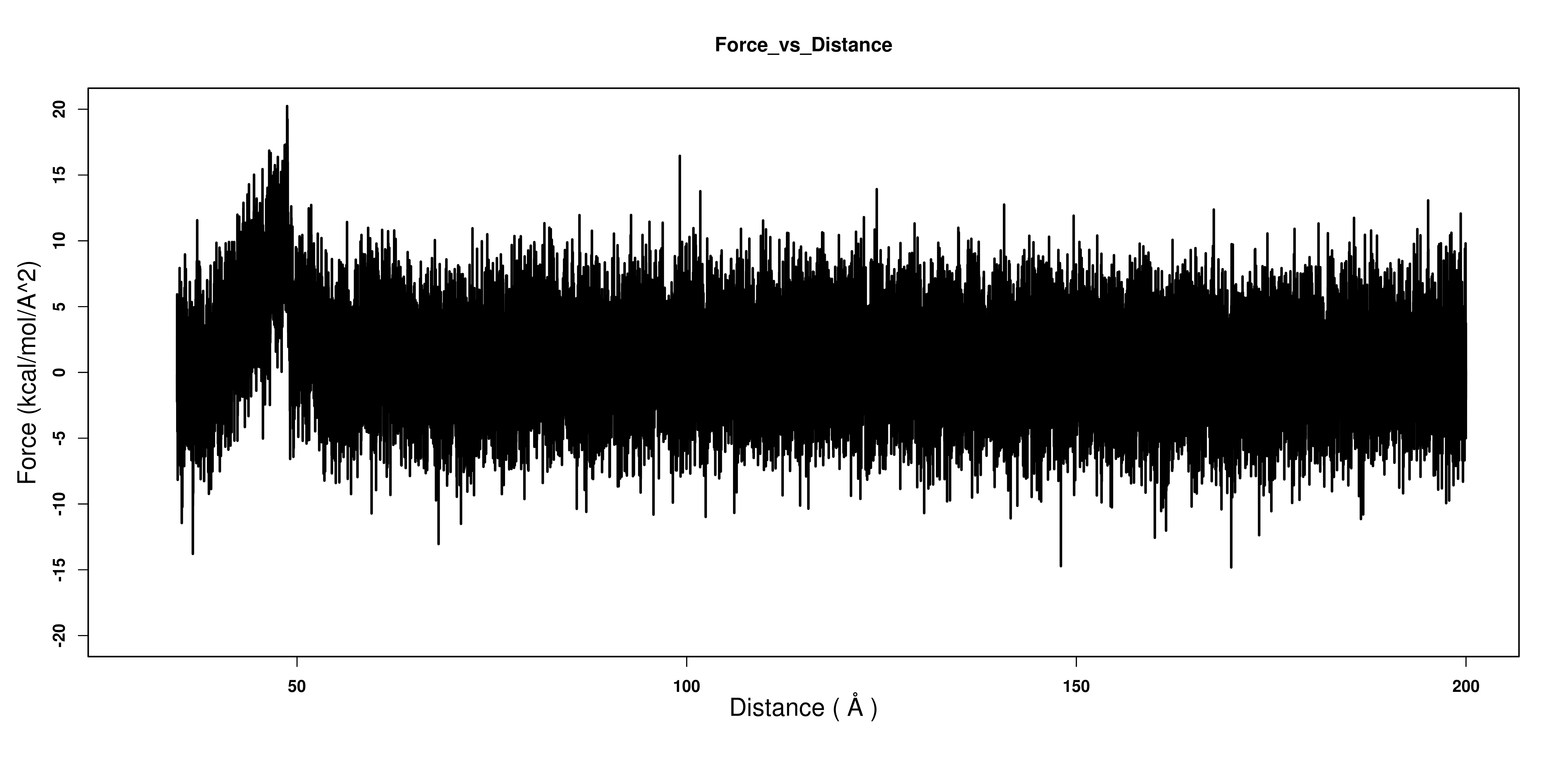
But I do not understand how to plot force vs Extension? Is it the column 3
vs column 1 of the DUMPAVG output file? When I plot this, it gives
something wrong for certain. The total force acting on the system
fluctuates around 0 (fig2).
How do obtain a force extension curve something like in the fig 3 (Obtained
from - https://doi.org/10.1002/prot.22314).
I am intrested in seeing the force profile corresponding to every secondary
structure as the protein unfolds.
Regards,
Aravind R
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Mon Oct 07 2019 - 00:30:02 PDT