Date: Tue, 17 Sep 2019 14:44:25 +0530
I am really very sorry sir, I miswrote the question.
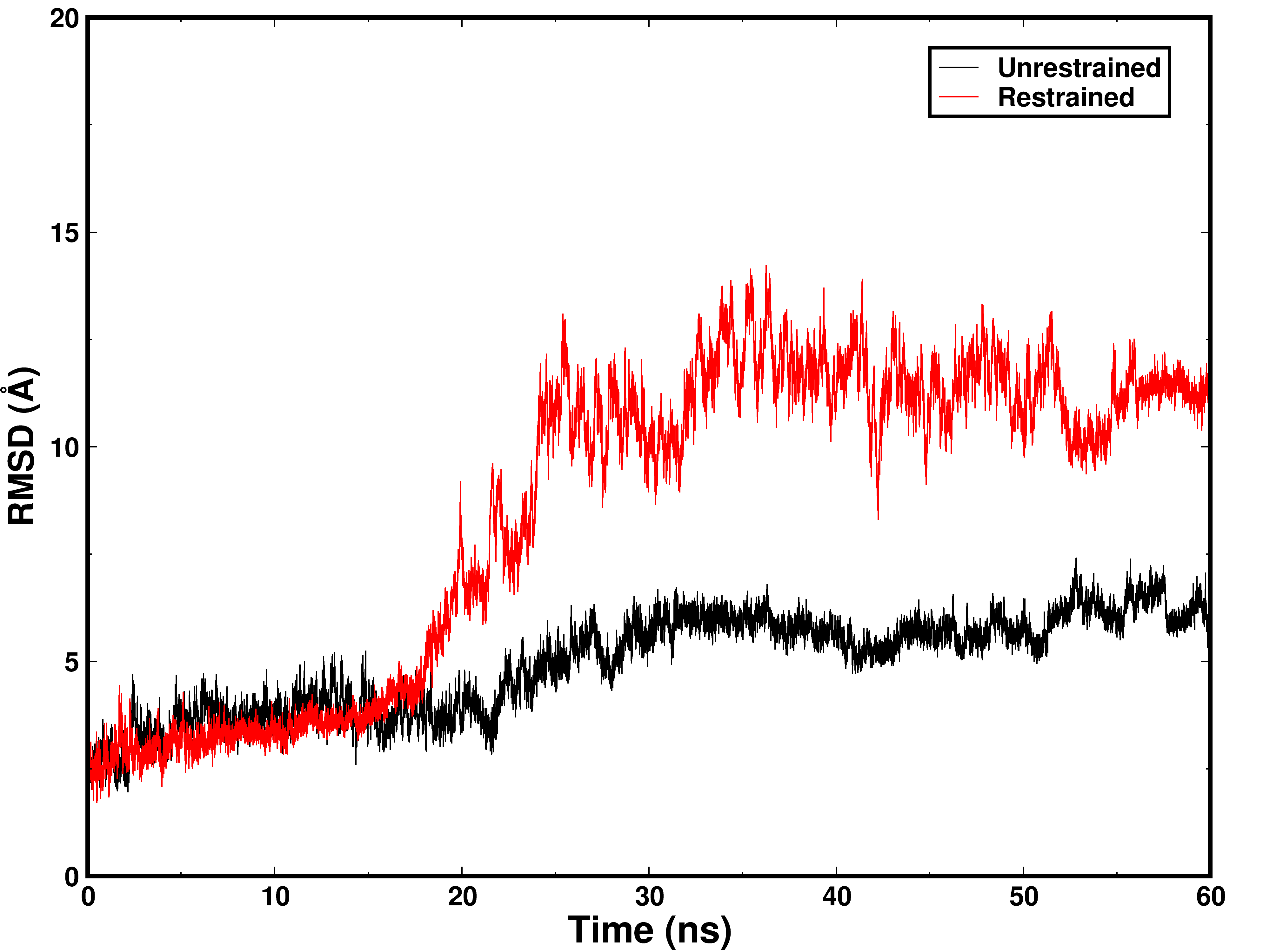
I found the RMSD value of the restrained system higher than the
unrestrained system, which is very unusual. I am also attaching the
result.
On Mon, Sep 16, 2019 at 6:57 PM Carlos Simmerling <
carlos.simmerling.gmail.com> wrote:
> Yes this is very sensible. I would expect that the restraints would keep
> the structure closer to the initial model (if that's what you use as the
> reference for rmsd), leading to lower rmsd with the restrained trajectory.
>
> On Mon, Sep 16, 2019, 9:20 AM Amit Kumar <ak543714.gmail.com> wrote:
>
> > Dear Amber users,
> > I ran a simulation of a fibrillar protein for few nanosecond (with and
> > without torsional restraint) using Amber16. I used implicit solvent
> (igb=5)
> > results when analysed in terms of RMSD, the RMSD value of the
> unrestrained
> > system was higher than the RMSD value of the restrained system. Is it
> > possible? Can you explain the phenomenon?
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: system_compare.png)
