Date: Tue, 30 Jul 2019 12:25:46 +0000
Dear Amber Users,
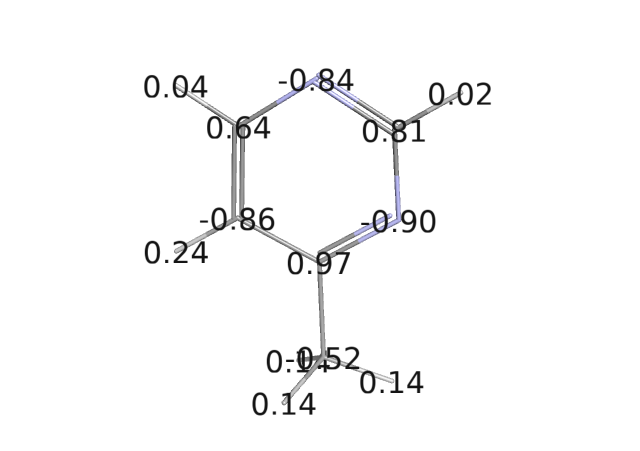
I would like to simulate a ligand-protein complex, so I calculated the charges RESP (from QM HF/6-31G* calculations) and I realized that one of the Carbon atom of a pyrimidine group in the ligand has a separation of charges at the C-H bond (in attachment a picture of a simplified system with the calculated charges). The C-H in position meta with respect of the 2 Nitrogen atoms of the pyrimidine ring has a polarized bond, C(-0.86)-H(0.24), in the same order of an O-H group of serine (taken from all_amino03.lib) , O(-0.64)-H(0.45). So, if I understand correct, this formed dipole could give, in principle, an Hydrogen bond interaction with a close by second dipole (at the proper distance and angle). Would the force field be able to describe it?
I searched the literature to understand how Hydrogen bonds are described in force fields but I could not find exactly this information. Can you please point me to some publications/information/literature.
Thanks in advance for any help,
Loris
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: pyrimidine_charges.png)
