Date: Fri, 19 Jul 2019 13:34:55 +0200
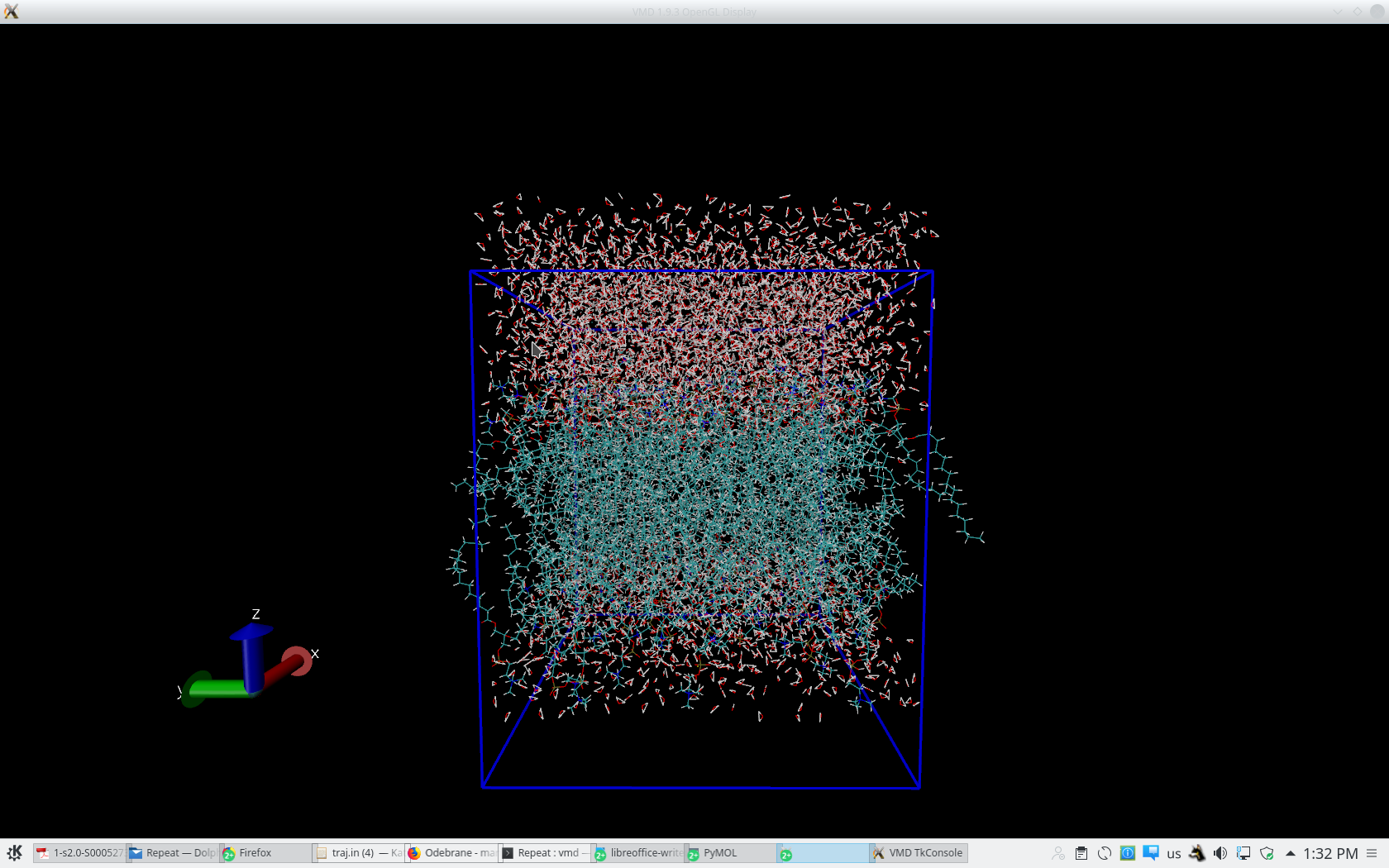
Indeed, the mask is ':POPC', but I still have some doubts when I visualize
it in VMD (you can see in the attached file how it looks with the pbc box)
cpptraj commands:
autoimage
center ":POPC"
Maria
pt., 19 lip 2019 o 11:13 Daniel Roe <daniel.r.roe.gmail.com> napisał(a):
> Hi,
>
> On Fri, Jul 19, 2019 at 4:17 AM Maria Bzówka <m.bzowka.tunnelinggroup.pl>
> wrote:
> > I assume that this is caused by the fact that CHARMM-GUI (which was used
> to
> > build the membrane) does not split the lipid into three "residues".
> > However, when I tried to use other mask definitions e.g. "POPC" or
> "MEMB" i
> > got the error:
>
> If you are using CHARMM membrane parameters, I suspect the mask you
> need is ':POPC'. Cpptraj actually has some useful commands you can use
> to figure out what things are named in a given topology file/test mask
> expressions; 'molinfo short', 'resinfo', 'atoms', etc. For example,
> below I will paste the output from cpptraj (run interactively) on a
> loaded PSF containing some proteins embedded in a lipid bilayer:
>
> > molinfo short
> [molinfo short]
> #Mol Count Natom Nres
> PHE 28 376 22
> POPC 525 134 1
> POPG 175 127 1
> TIP3 5950 3 1
> SOD 63 1 1
> > resinfo ^29
> [resinfo ^29]
> #Res Name First Last Natom #Orig #Mol
> 617 POPC 10529 10662 134 1 29
>
> So in this case, if I wanted to select the entire membrane I would use
> ':POPC,POPG' (or alternatively ':POP?').
>
> Hope this helps,
>
> -Dan
>
> >
> > AUTOIMAGE: To box center based on center of mass, anchor is first
> molecule.
> > [center "MEMB" origin]
> > CENTER: Centering coordinates using geometric center of atoms in mask
> > (*) to
> > coordinate origin.
> > Error: [center] Not all arguments handled: [ MEMB ]
> > 1 errors encountered reading input.
> >
> > So here my question is, how to define a mask properly so that it covers
> the
> > entire bilayer?
> >
> > Thank you in advance
> >
> > Maria
> >
> >
> >
> >
> > pon., 15 lip 2019 o 16:39 Daniel Roe <daniel.r.roe.gmail.com>
> napisał(a):
> >
> > > Hi,
> > >
> > > This is happening because autoimage is choosing a single molecule from
> > > one of the bilayers as the anchor. After "autoimage", you could
> > > 'center' on the bilayer (i.e. use 'center' and select the entire
> > > bilayer) and then image only water, ions with 'image bymol
> > > :WAT,Na+,Cl-' etc.
> > >
> > > Alternatively you could try to choose the end of one of the lipid
> > > chains (near the bilayer center) as the anchor instead of an entire
> > > lipid.
> > >
> > > Hope this helps,
> > >
> > > -Dan
> > >
> > > On Mon, Jul 15, 2019 at 9:53 AM Maria Bzówka <
> m.bzowka.tunnelinggroup.pl>
> > > wrote:
> > > >
> > > > Hi Dan,
> > > >
> > > > Thank you for your suggestion, it works. However, the new problem
> showed
> > > > up. Right now, the water layers do not have the same thickness (you
> can
> > > > see in the attached file). I don't know what to think about it.
> Does it
> > > > mean that something is wrong with the equilibration itself?
> > > >
> > > > Maria
> > > >
> > > > pon., 15 lip 2019 o 15:26 Daniel Roe <daniel.r.roe.gmail.com>
> > > napisał(a):
> > > >
> > > > > Hi,
> > > > >
> > > > > 'autoimage origin' will center the system at the coordinate origin
> > > > > (bottom corner of the box). Just omit the 'origin' keyword from the
> > > > > autoimage command and remove the subsequent center/image commands.
> > > > >
> > > > > Hope this helps,
> > > > >
> > > > > -Dan
> > > > >
> > > > > On Mon, Jul 15, 2019 at 7:32 AM Maria Bzówka <
> > > m.bzowka.tunnelinggroup.pl>
> > > > > wrote:
> > > > > >
> > > > > > Hi all,
> > > > > >
> > > > > > I work with the system which consists of 128 lipids bilayer and
> water
> > > > > > molecules. I used the cpptraj to center the whole system using
> > > following
> > > > > > commands:
> > > > > >
> > > > > > trajin step6.1_equilibration.nc
> > > > > > trajin step6.2_equilibration.nc
> > > > > > trajin step6.3_equilibration.nc
> > > > > > trajin step6.4_equilibration.nc
> > > > > > trajin step6.5_equilibration.nc
> > > > > > trajin step6.6_equilibration.nc
> > > > > > autoimage origin
> > > > > > center :1-128 mass origin
> > > > > > image origin center familiar
> > > > > >
> > > > > > In VMD the system itself looks fine, but when I wanted to check
> the
> > > pbc
> > > > > > box, it only partially overlaps with the system (you can see in
> the
> > > > > > attached file).
> > > > > > Should I somehow change the cpptraj commands?
> > > > > >
> > > > > > Thank you in advance for suggestions
> > > > > >
> > > > > > Maria
> > > > > > _______________________________________________
> > > > > > AMBER mailing list
> > > > > > AMBER.ambermd.org
> > > > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > > >
> > > > > _______________________________________________
> > > > > AMBER mailing list
> > > > > AMBER.ambermd.org
> > > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > > >
> > > > _______________________________________________
> > > > AMBER mailing list
> > > > AMBER.ambermd.org
> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: vmd3.png)
