Date: Tue, 30 Oct 2018 21:19:49 +0530
we can construct the input file required by * NAB* to build our 10-mer
polyA-polyT DNA duplex in the Arnott B-DNA canonical structure. This
program is given below. Basically, this is building two strands of A-T
paired DNA. For more specific information about the various options, see
the manual <http://ambermd.org/doc12/>.)
molecule m;
m = fd_helix( "abdna", "aaaaaaaaaa", "dna" );
putpdb( "nuc.pdb", m, "-wwpdb");
Put the above into a plain-text file called * nuc.nab
<http://ambermd.org/tutorials/basic/tutorial1/files/amber_input_files/nuc.nab>*.
*Note:* Before we run any of the programs provided with AMBER, we need to
make sure the Unix shell environment variable that specifies where AMBER is
installed is set properly. This is necessary for *xleap* and lets the
system know where the executables are located ($AMBERHOME/bin).
If you are running bash, csh or tcsh check the AMBERHOME environment
variable is set by typing:
echo $AMBERHOME
If you see:
AMBERHOME: Undefined variable.
(Using csh or tcsh) or simply a blank line (bash) then the AMBERHOME
environment variable was not set properly and you need to initialize it to
point to the AMBER installation directory. If AMBER is installed in
/usr/local/amber14 then you would type:
setenv AMBERHOME /usr/local/amber14
(csh or tcsh) or
export AMBERHOME=/usr/local/amber14
(bash).
While you are at it, you may want to update your .cshrc (csh), or
.bashrc (bash)
file to define this variable and add the AMBER binary directory to your
path (or list of directories searched for executables) at login. In your
.bash_login file [or the global /etc/bashrc file] (if using the bash
shell), add the following (substituting the correct path to AMBER):
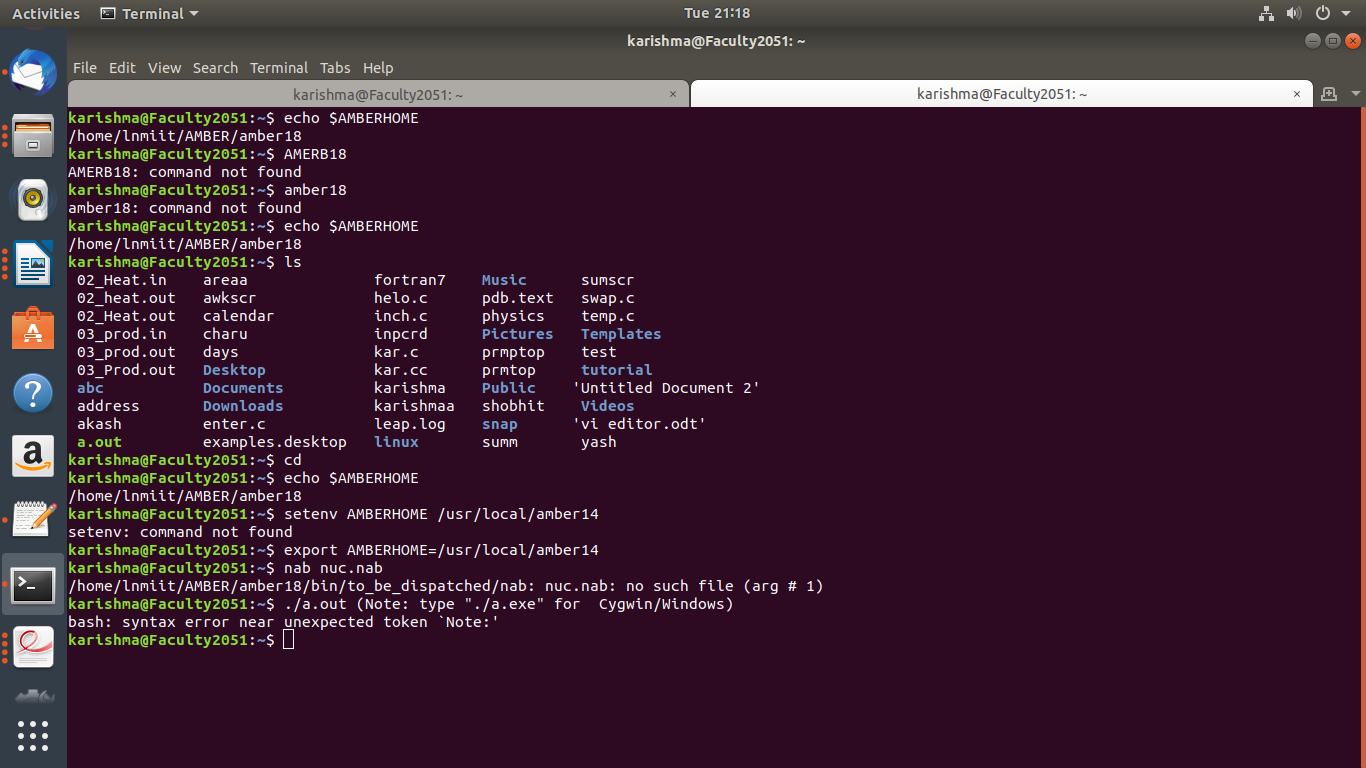
*sir i am getting the problem that after creating nuc.nab file in gedit it
is not exceuting i am sending you screen shot please help?*
On Wed, Oct 10, 2018 at 8:48 PM Elvis Martis <elvis.martis.bcp.edu.in>
wrote:
> HI,
> Once you have installed AMBER.
> type the following in your terminal
> source $AMBERHOME/amber.sh
> or
> source $AMBERHOME/amber.csh
> and then type
> xleap
>
> Best Regards
> Elvis Martis
> Mumbai, INDIA.
>
> ________________________________________
> From: Charu Sharma (JRF) <charu.sharma.lnmiit.ac.in>
> Sent: 10 October 2018 16:31
> To: amber.ambermd.org
> Subject: Re: [AMBER] Regarding Leap
>
> Hello sir,
> I want to know how to run the command of xleap in ubuntu while working in
> linux.
>
> On Tue, Oct 9, 2018 at 11:13 AM Elvis Martis <elvis.martis.bcp.edu.in>
> wrote:
>
> > Hi Charu/Karishma,
> > It is highly recommended that you read the AMBER manual (
> > http://ambermd.org/doc12/Amber18.pdf), and also, visit the amberMD web
> > page (http://ambermd.org/FileFormats.php) that has all your answers.
> > Here, you will find information on FF14SB (
> > http://pubs.acs.org/doi/abs/10.1021/acs.jctc.5b00255)
> > Leaprc.ff14SB is the file to source Amber14 force field parameters.
> > FYI, from AMBER16 onwards this is "leaprc.protein.ff14SB"
> >
> > Hope this helps
> >
> > Best Regards
> > Elvis Martis
> > Mumbai, INDIA.
> >
> > ________________________________________
> > From: Charu Sharma (JRF) <charu.sharma.lnmiit.ac.in>
> > Sent: 09 October 2018 10:03
> > To: amber-subscribe.ambermd.org; Dr. Ashok Garai; AMBER.ambermd.org
> > Subject: [AMBER] Regarding Leap
> >
> > Hello everyone,
> > This is karishma , i want to know basic difference between AMBER topology
> > file extension, coordinates file extension as well as about
> Leaparc.ff14SB.
> >
> > Thanking you,
> >
> >
> > Regards
> >
> > karishma Sharma
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2018-10-30_21-18-14.png)
