Date: Tue, 30 Oct 2018 10:59:25 +0000
Many thanks for your email Pr.Case,
My goal is to construct Polymer P3ht "C10H14S" (Degre of polymerization 50)
using Moltemplate ( is a general molecule builder and Force Field database
system for lammps), specifying coordinates, partial charges .. of each atom
of the monomer.
My idea is to create 3 Monomers :
- 2 Monomers of end chain ( attached to this mail )
- 1 Monomer (attached to this mail)
I had coordinates but still need to specify the partial charges of each
atom.
Le lun. 29 oct. 2018 à 12:10, David A Case <david.case.rutgers.edu> a
écrit :
> On Sun, Oct 28, 2018, soukaina louerdi wrote:
>
> > I use Avogadro to make a Molecular model (3-hexylthiophene* ( *C10H15S) )
> > and I get Pdb file (I have attached an image of the pdb file).
>
> Attaching the pdb file itself would be much more helpful, since we could
> try to reproduce the problem. But it looks like you are missing a
> hydrogen atom on a carbon atom in the ring.
>
> > Based on the Antechamber tutorial (sustiva) :
> > $AMBERHOME/bin/antechamber -i h_p3ht.pdb -fi pdb -o h_p3ht.mol2 -fo mol2
> > -nc 1 -m 2 -c bcc -s 2 -j 5
>
> The fact that you requested a multiplicity of 2 suggests that you
> intended to leave out the hydrogen, but gaff is designed for closed
> shell molecules. What is this ligand supposed to interact with? Does
> it polymerize with a second identical molecule? It's hard for me to
> figure out how you can have a odd number of electrons and yet a net
> charge of +1....but I could well be missing something from the
> screenshot you sent.
>
> > Warning: The unperturbed charge of the unit (-1.002000) is not zero.
>
> This is indeed odd, since you requested a net charge of +1, but got a
> net charge of -1.
>
> ....dac
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
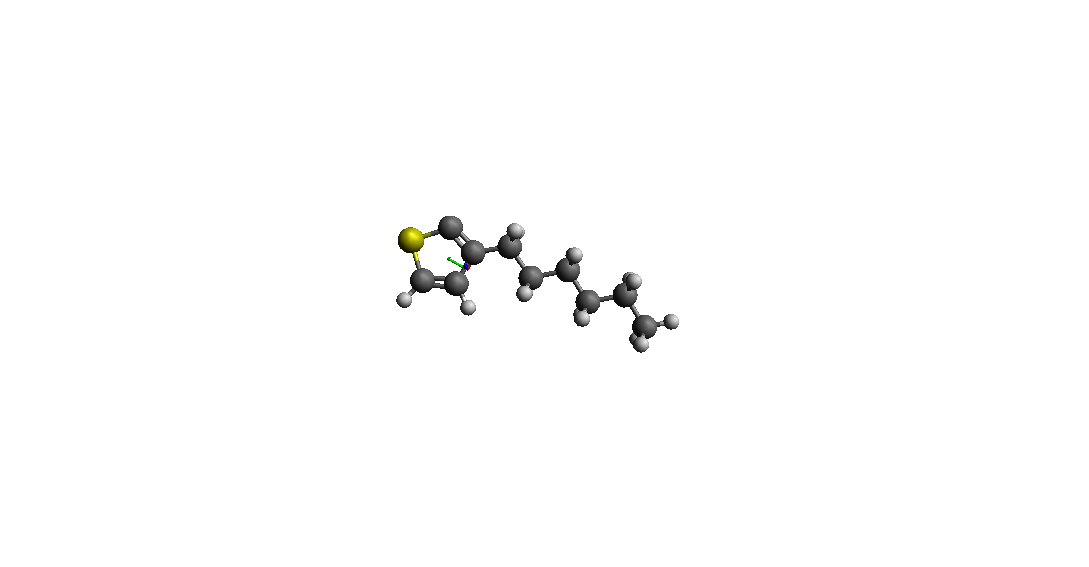
(image/png attachment: h_p3ht.png)
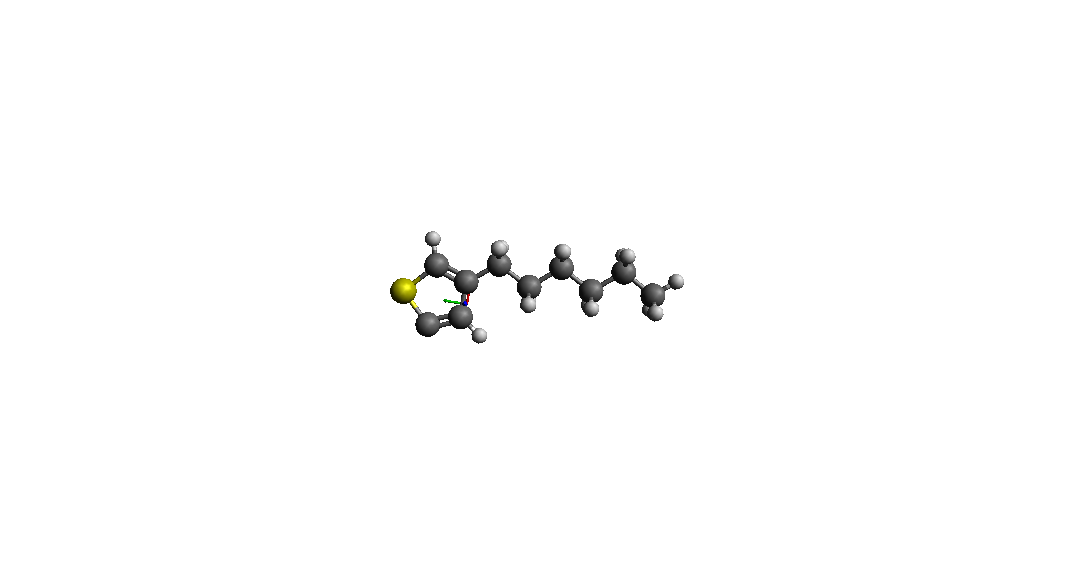
(image/png attachment: p3ht_h.png)
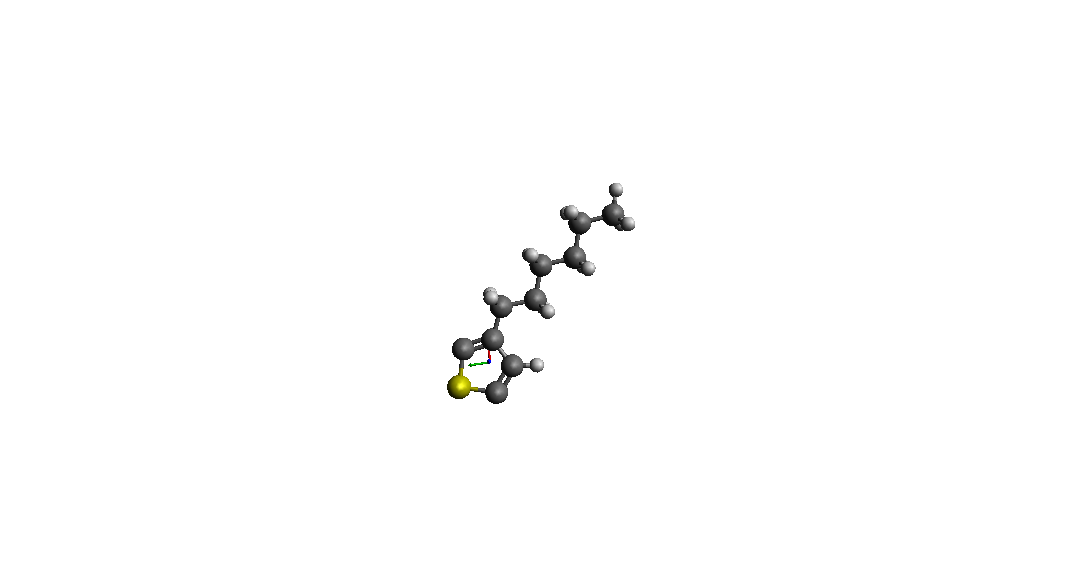
(image/png attachment: p3ht.png)
