Date: Wed, 10 Oct 2018 13:52:52 +0000
Sent from Mail<https://go.microsoft.com/fwlink/?LinkId=550986> for Windows 10
________________________________
From: fateme haghighi <fateme.haghighi.hotmail.com>
Sent: Wednesday, October 10, 2018 3:46:53 PM
To: AMBERLicensing.ucsf.edu
Subject: Fatal error in RESP calculation
Hi
I am using ambertools 18 to prepare topology of molecules I want to work with them.
I have used Gaussian 09 to calculate the charge with following detail:
#p sp b3lyp/6-31g* geom=connectivity scf=tight scf(maxcycle=800) pop=mk iop(6/33=2,6/42=6)
Even I have added iop(6/50=1) and I used the following command for RESP charge calculation:
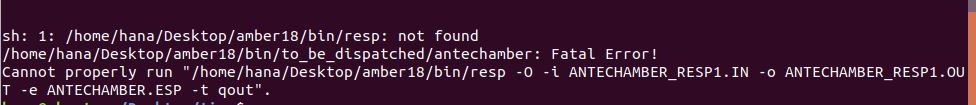
Antechamber –i tica.out –fi gout –o TIC.mol2 –fo mol2 –c resp –nc 0 –at amber –rn TIC
But when I have got the attached error. Would you please help me to solve the problem?
Thank you very much
Sent from Mail<https://go.microsoft.com/fwlink/?LinkId=550986> for Windows 10
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: antechamber_error1.JPG)
