Date: Wed, 25 Apr 2018 11:51:54 -0300
*Hello everyone, I'm a new user of Amber, and I'm experiencing some
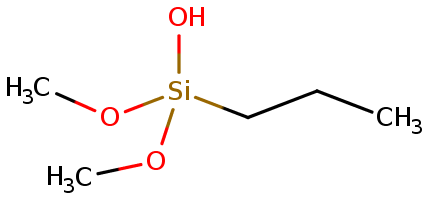
problems trying to obtain parameters for a siloxane unit (figure below),
using paramfit.*
*I'm interested in parametrize some dihedral, angles and bonds, mainly
involving Si, and the 4 atoms directly bonded in it (that were assigned
with different atom types). I’m considering as initial parameters,
analogous bonds, angles and dihedrals, changing Si for a c3 atom, and using
these values defined in gaff.dat.Lennard-Jones values for Si was obtained
from force field AMBER parm99, for docking
(https://users.rcc.uchicago.edu/~yadunand/swift-k/bgq-swift/dock/vdw_AMBER_parm99.defn
<https://users.rcc.uchicago.edu/~yadunand/swift-k/bgq-swift/dock/vdw_AMBER_parm99.defn>).Well,
I try to follow paramfit tutorial, but, for my system, i'm obtaining really
bad R^2 values for the first fitting of K (around 0.1), and for the
subsequently parameter fit (the best one was 0.66).I try different
approaches to solve this. 1) I improve my sampling, try 2 000 random
conformations (resulting 341 good conformations), and 20 000 random
conformations (resulting 3414 good conformations). In the last case, I find
the "best" result for R^2 in K fitting (0.1284) and at the actual parameter
fitting with R^2 = 0.6648.2) Another test was reducing the energy cutoff
for removing bad structures. Previous calculations consider a 2000 kcal/mol
cutoff, like in the tutorial. I reduce to 1500, cause some structures that
pass this minima are yet bad conformations (and gives error in Gaussian
calculation of single point, due overlapping atoms) -- and these ones are
removed from my valid_structures.mdcrd with a script that I wrote. Reducing
this cutoff shows worse results for R^2 (in both fitting K and parameters,
I tested this for a 2 000 conformation system).3) Changing the level of
theory. I started using b3lyp/6-31G* cause its the same level of theory
used for resp calculation of charges. So, I tried with a greater basis set,
6-311G*, and still using a hybrid functional, but now, PBE0. The results
were a little better, but, for 2 000 conformations, and 316 good ones
(energy cutoff of 2000 kcal/mol), the fitting o K shows R^2 of 0.1034, and
for the parameters R^2 = 0.6378.4) I tried to see the plots using
plot_energy.x and scatterplots.sh, as the tutorial teach. Try to change the
weight of some structures, but the R^2 gets even worse. It was difficult
due the high number of conformations involved.- My QM calculations were
just single points, I need to use a different approach? - There are some
warnings about the parameters, that I try to solve by increasing the
sampling, but this warnings are always present.How can I improve my
fitting? Cause I don't have any other ideas to solve this and get a better
fit. Thanks in advantage!Here are some informations about my system:*Atom
types: (I’m using gaff)SI - Si atomJ3 - c3 bonded to SiQH - oh bonded to
SiQS - os bonded to Si*My parameters to fit:# BOND INFORMATION:#### BOND
REQ KR #### J3 hc 1 1 QH ho 1 1 c3
J3 1 1 J3 SI 1 1 SI QH 1 1 SI QS 1
1 QS c3 1 1## ANGLE PARAMETERS:#### ANGLE KT THEQ
#### c3 J3 hc 1 1 J3 c3 hc 1 1 hc J3 SI
1 1 hc J3 hc 1 1 SI QH ho 1 1 QS
c3 h1 1 1 c3 c3 J3 1 1 c3 J3 SI 1 1
J3 SI QH 1 1 J3 SI QS 1 1 SI QS c3
1 1 QH SI QS 1 1 QS SI QS 1 1## DIHEDRAL
PARAMETERS:#### DIHEDRAL TERM KP NP PHASE #### c3 c3 J3 hc
0 1 1 1 J3 SI QH ho 0 1 1 1 J3 SI QH ho
1 1 1 1 hc c3 J3 hc 0 1 1 1 hc c3 J3 SI
0 1 1 1 hc J3 SI QH 0 1 1 1 hc J3 SI QH
1 1 1 1 hc J3 SI QS 0 1 1 1 hc J3 SI QS
1 1 1 1 SI QS c3 h1 0 1 1 1 QS SI QH ho
0 1 1 1 c3 c3 J3 SI 0 1 1 1 c3 c3 J3 SI
1 1 1 1 c3 c3 J3 SI 2 1 1 1 c3 J3 SI QH
0 1 1 1 c3 J3 SI QS 0 1 1 1 J3 SI QS c3
0 1 1 1 J3 SI QS c3 1 1 1 1 QH SI QS c3
0 1 1 1 QS SI QS c3 0 1 1 1 QS SI QS c3
1 1 1 1 QS SI QS c3 2 1 1 1*Outputs for a
fitting with 20 000 structures, and 3179 good ones (cutoff of 2000 kcal/mol
results in 3414 “good” structures, but 235 presented error reported
previously, and were removed from valid.mdcrd)*Fitting K:
*****************************************************
* AMBER Bond Angle and Dihedral Parameter Optimiser *
* * *
v3.0.0 * * *
* Written by: * * Robin Betz
(2011) * * Ross Walker (2004)
* * The Walker Molecular Dynamics Lab
* * University of California, San Diego *
* La Jolla, California, 92092 * *
USA *
******************************************************************************************************************************************
Execution started at:|
Tue Apr 24 16:00:09 2018|| Random seed =
1524596409 Reading job control file: fit_K_sil.in Job Control: Read a total
of 12 lines from job_control file. 6 options set. Reading prmtop file :
sil.prmtop Prmtop (unique): Found 10 unique bonds. Prmtop (unique):
Found 16 unique angles. Prmtop (unique): Found 16 unique dihedrals.
Prmtop (unique): Found 24 unique dihedral terms. Reading mdcrd file:
sil_valid_structures.mdcrd Reading mdcrd file :
sil_valid_structures.mdcrd Coordinate file passed format check Reading
energy file or directory : energy_qm_sil.dat
---------------------------------------------------------------------
| OPTIONS SUMMARY | |
--------------- | | Summary of Run
Type Options: | | Run Mode =
FIT, Minimiser = SIMPLEX | | Function to
be Fit = SUM_SQUARES_AMBER_STANDARD | |
| | Terms
to be fit: | | K =
1, UNIQUE_BONDS = 10, UNIQUE ANGLES = 16 | |
UNIQUE DIHEDRALS = 16 TOTAL DIHEDRAL TERMS = 24 | |
NBONDS = 22, NANGLES = 39, NDIHEDRALS = 57 | |
Total dimensions of fit = 1 | |
| |
Sample structures for least squares fit = 3179 |
| |
| |
| Energy Correction Term (K) = TO BE FIT
| |
| |
BONDFC_dx = 5.0000 | | BONDEQ_dx =
0.0200 | | ANGLEFC_dx = 1.0000
| | ANGLEEQ_dx = 0.0500
| | DIHEDRALBH_dx = 0.2000 |
| DIHEDRALN_dx = 0.0100 | | DIHEDRALG_dx =
0.0500 | | K_dx = 10.0000
| |
| |
Convergence requested to within 1.0000E-15 |
| |
| Estimate Memory Usage (per cpu):
| | Coordinate info will be read
from disk as required. | | OPTION STORAGE = 67
bytes | | PRMTOP STORAGE = 389242
bytes | | COORDINATE STORAGE = 1958304
bytes | | SIMPLEX ARRAY STORAGE = 168 bytes
| |
| | TOTAL
ESTIMATED MEMORY USAGE = 2347781 bytes | |
|
---------------------------------------------------------------------
* Input structures passed dihedral span check.
------------------------------- INITIAL PARAMETERS
-------------------------------- Parameters for force field equation:
AMBER_STANDARD: (* means parameter is NOT constant during fit)
*K = 0.000000 kcal/mol (hc-c3) Kr =
330.6000 kcal/(mol A)^2, r_eq = 1.0969 A (J3-hc) Kr =
330.6000 kcal/(mol A)^2, r_eq = 1.0969 A (QH-ho) Kr =
371.4000 kcal/(mol A)^2, r_eq = 0.9730 A (c3-h1) Kr =
330.6000 kcal/(mol A)^2, r_eq = 1.0969 A (c3-c3) Kr =
300.9000 kcal/(mol A)^2, r_eq = 1.5375 A (c3-J3) Kr =
300.9000 kcal/(mol A)^2, r_eq = 1.5375 A (J3-SI) Kr =
206.0000 kcal/(mol A)^2, r_eq = 1.7800 A (SI-QH) Kr =
203.2000 kcal/(mol A)^2, r_eq = 1.7500 A (SI-QS) Kr =
599.8000 kcal/(mol A)^2, r_eq = 1.7580 A (QS-c3) Kr =
308.6000 kcal/(mol A)^2, r_eq = 1.4316 A (hc-c3-c3) Kt = 46.3400
kcal/(mol rad)^2, th_eq = 109.8000 deg (hc-c3-hc) Kt = 39.4000
kcal/(mol rad)^2, th_eq = 107.5800 deg (c3-J3-hc) Kt = 46.3400
kcal/(mol rad)^2, th_eq = 109.8000 deg (J3-c3-hc) Kt = 46.3400
kcal/(mol rad)^2, th_eq = 109.8000 deg (hc-J3-SI) Kt = 46.3400
kcal/(mol rad)^2, th_eq = 109.8000 deg (hc-J3-hc) Kt = 39.4000
kcal/(mol rad)^2, th_eq = 107.5800 deg (SI-QH-ho) Kt = 47.3800
kcal/(mol rad)^2, th_eq = 107.2600 deg (QS-c3-h1) Kt = 50.8000
kcal/(mol rad)^2, th_eq = 109.7800 deg (h1-c3-h1) Kt = 39.2400
kcal/(mol rad)^2, th_eq = 108.4600 deg (c3-c3-J3) Kt = 62.8600
kcal/(mol rad)^2, th_eq = 111.5100 deg (c3-J3-SI) Kt = 62.8600
kcal/(mol rad)^2, th_eq = 111.5100 deg (J3-SI-QH) Kt = 67.4700
kcal/(mol rad)^2, th_eq = 110.1900 deg (J3-SI-QS) Kt = 68.0000
kcal/(mol rad)^2, th_eq = 107.9700 deg (SI-QS-c3) Kt = 62.7000
kcal/(mol rad)^2, th_eq = 112.4800 deg (QH-SI-QS) Kt = 72.5800
kcal/(mol rad)^2, th_eq = 109.3800 deg (QS-SI-QS) Kt = 72.7400
kcal/(mol rad)^2, th_eq = 108.2900 deg (hc-c3-c3-J3) Kp = 0.1556
kcal/mol, Np = 3.0000, Phase = 0.0000 Deg (hc-c3-c3-hc) Kp =
0.1500 kcal/mol, Np = 3.0000, Phase = 0.0000 Deg (c3-c3-J3-hc) Kp
= 0.1600 kcal/mol, Np = 3.0000, Phase = 0.0000 Deg (J3-SI-QH-ho)
Kp = 0.2500 kcal/mol, Np = 1.0000, Phase = 0.0000 Deg
(J3-SI-QH-ho) Kp = 0.1600 kcal/mol, Np = 3.0000, Phase = 0.0000
Deg (hc-c3-J3-hc) Kp = 0.1500 kcal/mol, Np = 3.0000, Phase =
0.0000 Deg (hc-c3-J3-SI) Kp = 0.1600 kcal/mol, Np = 3.0000, Phase
= 0.0000 Deg (hc-J3-SI-QH) Kp = 0.2500 kcal/mol, Np = 1.0000,
Phase = 0.0000 Deg (hc-J3-SI-QH) Kp = 0.0000 kcal/mol, Np =
3.0000, Phase = 0.0000 Deg (hc-J3-SI-QS) Kp = 0.2500 kcal/mol, Np
= 1.0000, Phase = 0.0000 Deg (hc-J3-SI-QS) Kp = 0.0000 kcal/mol,
Np = 3.0000, Phase = 0.0000 Deg (SI-QS-c3-h1) Kp = 0.3833
kcal/mol, Np = 3.0000, Phase = 0.0000 Deg (QS-SI-QH-ho) Kp =
0.1667 kcal/mol, Np = 3.0000, Phase = 0.0000 Deg (c3-c3-J3-SI) Kp
= 0.2000 kcal/mol, Np = 1.0000, Phase = 180.0001 Deg (c3-c3-J3-SI)
Kp = 0.2500 kcal/mol, Np = 2.0000, Phase = 180.0001 Deg
(c3-c3-J3-SI) Kp = 0.1800 kcal/mol, Np = 3.0000, Phase = 0.0000
Deg (c3-J3-SI-QH) Kp = 0.1556 kcal/mol, Np = 3.0000, Phase =
0.0000 Deg (c3-J3-SI-QS) Kp = 0.1556 kcal/mol, Np = 3.0000, Phase
= 0.0000 Deg (J3-SI-QS-c3) Kp = 0.1000 kcal/mol, Np = 2.0000,
Phase = 180.0001 Deg (J3-SI-QS-c3) Kp = 0.3800 kcal/mol, Np =
3.0000, Phase = 0.0000 Deg (QH-SI-QS-c3) Kp = 0.3833 kcal/mol, Np
= 3.0000, Phase = 0.0000 Deg (QS-SI-QS-c3) Kp = 1.3500 kcal/mol,
Np = 1.0000, Phase = 180.0001 Deg (QS-SI-QS-c3) Kp = 0.8500
kcal/mol, Np = 2.0000, Phase = 180.0001 Deg (QS-SI-QS-c3) Kp =
0.1000 kcal/mol, Np = 3.0000, Phase = 0.0000 Deg
-----------------------------------------------------------------------------------
Sum of squares for initial parameters = 639507927287609.5000000000
kcal^2/mol^2 R^2 value for initial parameters = 0.128447
Calculated energy with initial parameters for structure 1 = 1755.187168
KCal/mol Actual energy for structure 1 should be =
-445806.454765 KCal/mol --------------------------------- SIMPLEX
MINIMISATION ---------------------------- Minimising function
SUM_SQUARES_AMBER_STANDARD, using the SIMPLEX METHOD
-------------------------------------- CONVERGENCE
-------------------------------- Step 0: Conv= 4.2670E-05
min=639507927287609.5000,max=639535215666118.6250 avg639521571476864.0000
Step 1: Conv= 5.7680E-03 min=1226027881.6924 max=1233120095.5567
avg=1229573988.6245 Step 2: Conv= 0.0000E+00 min=1210774894.5474
max=1210774894.5474 avg=1210774894.5474
-----------------------------------------------------------------------------------
Convergence ratio of 0.0000E+00 is better than convergence criteria
of 1.0000E-15. Function Converged - Total function evaluations = 107
Convergence to 1.0000E-15 in Simplex routine achieved after 50 cycles.
(25 INNER x 2 OUTER CYCLES) ------------------------------- FINAL
PARAMETERS --------------------------------- Parameters for force field
equation: AMBER_STANDARD: (* means parameter is NOT constant during fit)
*K = -448515.329474 kcal/mol (hc-c3) Kr
= 330.6000 kcal/(mol A)^2, r_eq = 1.0969 A (J3-hc) Kr =
330.6000 kcal/(mol A)^2, r_eq = 1.0969 A (QH-ho) Kr =
371.4000 kcal/(mol A)^2, r_eq = 0.9730 A (c3-h1) Kr =
330.6000 kcal/(mol A)^2, r_eq = 1.0969 A (c3-c3) Kr =
300.9000 kcal/(mol A)^2, r_eq = 1.5375 A (c3-J3) Kr =
300.9000 kcal/(mol A)^2, r_eq = 1.5375 A (J3-SI) Kr =
206.0000 kcal/(mol A)^2, r_eq = 1.7800 A (SI-QH) Kr =
203.2000 kcal/(mol A)^2, r_eq = 1.7500 A (SI-QS) Kr =
599.8000 kcal/(mol A)^2, r_eq = 1.7580 A (QS-c3) Kr =
308.6000 kcal/(mol A)^2, r_eq = 1.4316 A (hc-c3-c3) Kt = 46.3400
kcal/(mol rad)^2, th_eq = 109.8000 deg (hc-c3-hc) Kt = 39.4000
kcal/(mol rad)^2, th_eq = 107.5800 deg (c3-J3-hc) Kt = 46.3400
kcal/(mol rad)^2, th_eq = 109.8000 deg (J3-c3-hc) Kt = 46.3400
kcal/(mol rad)^2, th_eq = 109.8000 deg (hc-J3-SI) Kt = 46.3400
kcal/(mol rad)^2, th_eq = 109.8000 deg (hc-J3-hc) Kt = 39.4000
kcal/(mol rad)^2, th_eq = 107.5800 deg (SI-QH-ho) Kt = 47.3800
kcal/(mol rad)^2, th_eq = 107.2600 deg (QS-c3-h1) Kt = 50.8000
kcal/(mol rad)^2, th_eq = 109.7800 deg (h1-c3-h1) Kt = 39.2400
kcal/(mol rad)^2, th_eq = 108.4600 deg (c3-c3-J3) Kt = 62.8600
kcal/(mol rad)^2, th_eq = 111.5100 deg (c3-J3-SI) Kt = 62.8600
kcal/(mol rad)^2, th_eq = 111.5100 deg (J3-SI-QH) Kt = 67.4700
kcal/(mol rad)^2, th_eq = 110.1900 deg (J3-SI-QS) Kt = 68.0000
kcal/(mol rad)^2, th_eq = 107.9700 deg (SI-QS-c3) Kt = 62.7000
kcal/(mol rad)^2, th_eq = 112.4800 deg (QH-SI-QS) Kt = 72.5800
kcal/(mol rad)^2, th_eq = 109.3800 deg (QS-SI-QS) Kt = 72.7400
kcal/(mol rad)^2, th_eq = 108.2900 deg (hc-c3-c3-J3) Kp = 0.1556
kcal/mol, Np = 3.0000, Phase = 0.0000 Deg (hc-c3-c3-hc) Kp =
0.1500 kcal/mol, Np = 3.0000, Phase = 0.0000 Deg *
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
