Date: Mon, 22 Jan 2018 17:03:43 +0530
Dear Sir
I have change name from HEM to FE in PDB file. now its working.
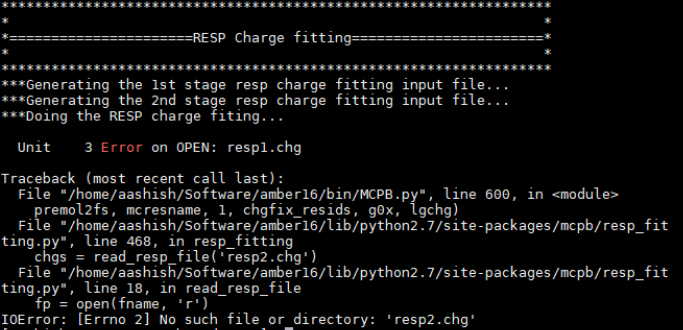
but i am getting error in 3rd step MCPB.py -i mcpb_3rd.in -s 3
IOError: [Errno 2]no such file or directory: 'resp2.chg'
I have searched on amber forum but this error is not answered .
Regards
Aashish Bhatt
On Wed, Jan 17, 2018 at 1:37 AM, Pengfei Li <ambermailpengfei.gmail.com>
wrote:
> Hi Aashish,
>
> Do you have the FE.mol2 file exactly equal to the FE.mol2 file in this
> tutorial: http://ambermd.org/tutorials/advanced/tutorial20/mcpbpy_heme.htm
> <http://ambermd.org/tutorials/advanced/tutorial20/mcpbpy_heme.htm>? That
> could be the reason.
>
> Kind regards,
> Pengfei
>
> > On Jan 8, 2018, at 10:47 PM, Aashish Bhatt <aashish.ph16221.inst.ac.in>
> wrote:
> >
> > Dear Sir
> >
> > I am preparing force field parameter of HEME so I am following the
> tutorial
> > (*http://ambermd.org/tutorials/advanced/tutorial20/mcpbpy_heme.htm
> > <http://ambermd.org/tutorials/advanced/tutorial20/mcpbpy_heme.htm>)*
> and I
> > have found the KeyError: 'FE' .All the required files are created but
> > file_small.res is empty.,so am unable to understand the results and the
> > error in my calculations if any . I will be highly grateful if you could
> > help me reading the error attached below .
> >
> > $ MCPB.py -i 4lod.in -s 1
> >
> > ******************************************************************
> > * Welcome to use the MCPB.py program *
> > * Version 2.0 *
> > * Author: Pengfei Li *
> > * Merz Research Group *
> > * Michigan State University *
> > ******************************************************************
> > The input file you are using is : 4lod.in
> >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> > The following is the input variable you have:
> > The variable ion_ids is : [23399]
> > The variable ion_info is : []
> > The variable ion_mol2files is : ['FE.mol2']
> > The variable original_pdb is : 4lod_add_H.pdb
> > The variable add_bonded_pairs is : []
> > The variable additional_resids is : []
> > The variable anglefc_avg is : 0
> > The variable bondfc_avg is : 0
> > The variable cut_off is : 2.8
> > The variable chgfix_resids is : []
> > The variable force_field is : ff14SB
> > The variable frcmodfs is : ['HEM.frcmod']
> > The variable gaff is : 1
> > The variable group_name is : 4lod
> > The variable ion_paraset is : CM (Only for nonbonded model)
> > The variable large_opt is : 0
> > The variable lgmodel_chg is : -99
> > -99 means program will assign a charge automatically.
> > The variable naa_mol2files is : ['HEM.mol2']
> > The variable scale_factor is : 1.0
> > ATTENTION: This is the scale factor of frequency. The
> > force constants will be scaled by multiplying the square
> > of scale_factor.
> > The variable smmodel_chg is : -99
> > -99 means program will assign a charge automatically.
> > The variable software_version is : g09
> > The variable sqm_opt is : 0
> > The variable water_model is : TIP3P
> > ******************************************************************
> > * *
> > *=======================Metal Site Information===================*
> > * *
> > ******************************************************************
> > ***Selected Metal ion FE is atom 23399 in residue 1495-HEM
> > 12-CYS.SG is in 2.8 Angstrom of or set bonded (in the input file) to
> (one
> > of) these metal ions
> > 25-HIE.NE2 is in 2.8 Angstrom of or set bonded (in the input file) to
> (one
> > of) these metal ions
> > 1494-HEM.NA is in 2.8 Angstrom of or set bonded (in the input file) to
> (one
> > of) these metal ions
> > 1494-HEM.NB is in 2.8 Angstrom of or set bonded (in the input file) to
> (one
> > of) these metal ions
> > 1494-HEM.NC is in 2.8 Angstrom of or set bonded (in the input file) to
> (one
> > of) these metal ions
> > 1494-HEM.ND is in 2.8 Angstrom of or set bonded (in the input file) to
> (one
> > of) these metal ions
> > ***The following residues are in the Metal Site:
> > Residue 12-CYS
> > Residue 25-HIE
> > Residue 1494-HEM
> > Residue 1495-HEM
> > ***The small model contains the following residues:
> > [12, 25, 1494, 1495]
> > ***The large model contains the following residues:
> > [11, 12, 13, 24, 25, 26, 1494, 1495]
> > ******************************************************************
> > * *
> > *=======================Building models==========================*
> > * *
> > ******************************************************************
> > ***Creating the small model...
> > It contains the residue 12-CYS as sidechain coordinated.
> > It contains the residue 25-HIE as sidechain coordinated.
> > It contains the residue 1494-HEM as normal.
> > It contains the residue 1495-HEM as normal.
> > Totally there are 97 atoms in the small model.
> > Totally there are 389 electrons in the small model.
> > ***Creating the standard model...
> > It contains the residue 12-CYS as normal.
> > It contains the residue 25-HIE as normal.
> > It contains the residue 1494-HEM as normal.
> > It contains the residue 1495-HEM as normal.
> > Totally there are 101 atoms in the standard model.
> > ***Creating the large model...
> > Creating the residue 11-ARG into ACE...
> > It contains the residue 12-CYS as normal.
> > Creating the residue 13-THR into NME...
> > Creating the residue 24-PRO into ACE...
> > It contains the residue 25-HIE as normal.
> > Creating the residue 26-HIE into NME...
> > It contains the residue 1494-HEM as normal.
> > It contains the residue 1495-HEM as normal.
> > Totally there are 125 atoms in the large model.
> > Totally there are 509 electrons in the large model.
> > Traceback (most recent call last):
> > File "/home/ehesan/Software/amber16/bin/MCPB.py", line 556, in <module>
> > premol2fs, cutoff, watermodel, 0, largeopt, sqmopt, smchg, lgchg)
> > File
> > "/home/Software/amber16/lib/python2.7/site-packages/mcpb/
> gene_model_files.py",
> > line 1745, in gene_model_files
> > ionids, chargedict, lgchg, outf, watermodel, largeopt, sqmopt)
> > File
> > "/home/Software/amber16/lib/python2.7/site-packages/mcpb/
> gene_model_files.py",
> > line 1441, in build_large_model
> > chargedict, IonLJParaDict, largeopt)
> > File
> > "/home/Software/amber16/lib/python2.7/site-packages/msmtmol/gauio.py",
> line
> > 156, in write_gau_mkf
> > chg = int(round(chargedict[ionname], 0))
> > KeyError: 'FE'
> >
> >
> >
> >
> >
> > Thanking You;
> > With Regards;
> > Aashish Bhatt (PhD scholar ,India)
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: error.png)
