Date: Fri, 19 Jan 2018 16:25:22 +0000 (UTC)
Dear Dan
We decided to increase the box size.
When we try 'solvatebox prot TIP3PBOX 10 iso', the structure is very weird as you can see in the attached pic, this didn't happen without iso
For 'solvateoct prot TIP3PBOX 10 iso' it gives the same result without iso
If I want to use the octahedral one with 10 A, but with less cut on the edges (equal dimentions and faces) what shall I do? Kind Regards,Ahmed
From: Daniel Roe <daniel.r.roe.gmail.com>
To: AMBER Mailing List <amber.ambermd.org>
Sent: Wednesday, January 17, 2018 4:55 PM
Subject: Re: [AMBER] Reorienting the protein during simulation
Hi,
On Wed, Jan 17, 2018 at 5:39 AM, Ahmed Mashaly <mashaly_1988.yahoo.com> wrote:
> The box fits very well if the protein is aligned to the main axis, the problem happens after it rotates 90 degrees (it is a very tall protein, so it exceeds the box sides when it is rotated) and one side may interact with the other side in case of periodic conditions.
> Another problem is that we are expecting a change in conformation, so we don't want to apply restraints, other solution is to increase the box size from 14 Angstrom to 20 which will take double of simulation time. So before moving to this step I want to make sure that there is no other solution through cpptraj actions?
When your protein rotates with respect to the unit cell axes there is
no "fixing" that. If you try to rotate the system to make it look
better then you're putting the system out of alignment with the
original unit cell vectors and imaging breaks. Why do you want to
reorient it at all?
-Dan
>
>
>
> From: Daniel Roe <daniel.r.roe.gmail.com>
> To: AMBER Mailing List <amber.ambermd.org>
> Sent: Tuesday, January 16, 2018 3:54 PM
> Subject: Re: [AMBER] Reorienting the protein during simulation
>
> Hi,
>
> The important thing to remember is that with periodic boundary
> conditions your protein is *never* really outside the box. It may
> appear to be outside the boundaries of your unit cell, but it is
> always interacting with its images. If your protein is rotating so
> that it is no longer aligned with your unit cell axes it is because it
> is favorable to do so. Your box may be too small. If you absolutely
> need the protein to remain aligned with e.g. the Z axis you'll
> probably have to apply some external restraint and make sure you
> correct for it in any later analysis if necessary.
>
> On Tue, Jan 16, 2018 at 5:52 AM, Ahmed Mashaly <mashaly_1988.yahoo.com> wrote:
>> This is what I use for recentering>
>> trajin my_rst_file.r>
>> autoimage
>> center: 1-1500 mass origin>
>> image origin center
>
> The 'center' and 'image' commands in this case will override whatever
> was done with 'autoimage', so it's not useful to combine the two.
> Either use 'autoimage' (recommended) *or* use 'image'/'center'.
>
>> I also tried to add>
>> prinicipal :1-1500 dorotation mass
>
> Note that the rotation done by 'principal' means that further imaging
> after this will not be possible.
>
> -Dan
>
> --
> -------------------------
> Daniel R. Roe
> Laboratory of Computational Biology
> National Institutes of Health, NHLBI
> 5635 Fishers Ln, Rm T900
> Rockville MD, 20852
> https://www.lobos.nih.gov/lcb
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
-- ------------------------- Daniel R. Roe Laboratory of Computational Biology National Institutes of Health, NHLBI 5635 Fishers Ln, Rm T900 Rockville MD, 20852 https://www.lobos.nih.gov/lcb _______________________________________________ AMBER mailing list AMBER.ambermd.org http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
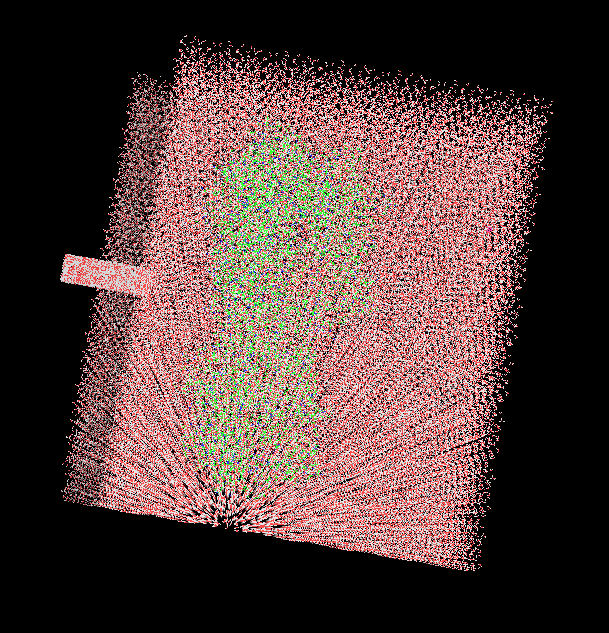
(image/png attachment: box_error.png)
(image/png attachment: oct.png)
