Date: Thu, 16 Nov 2017 16:12:27 +0100
Dear amber users,
I have been trying to run simple explicit solvent simulation on a Amber14
machine while using AmberTools16 generated input files.
I generate the topology and run Minimization and heating steps and finally
submit the job to the Amber14 machine. Also, my peptide contains
non-standard residues like AIB and D-isovaline.
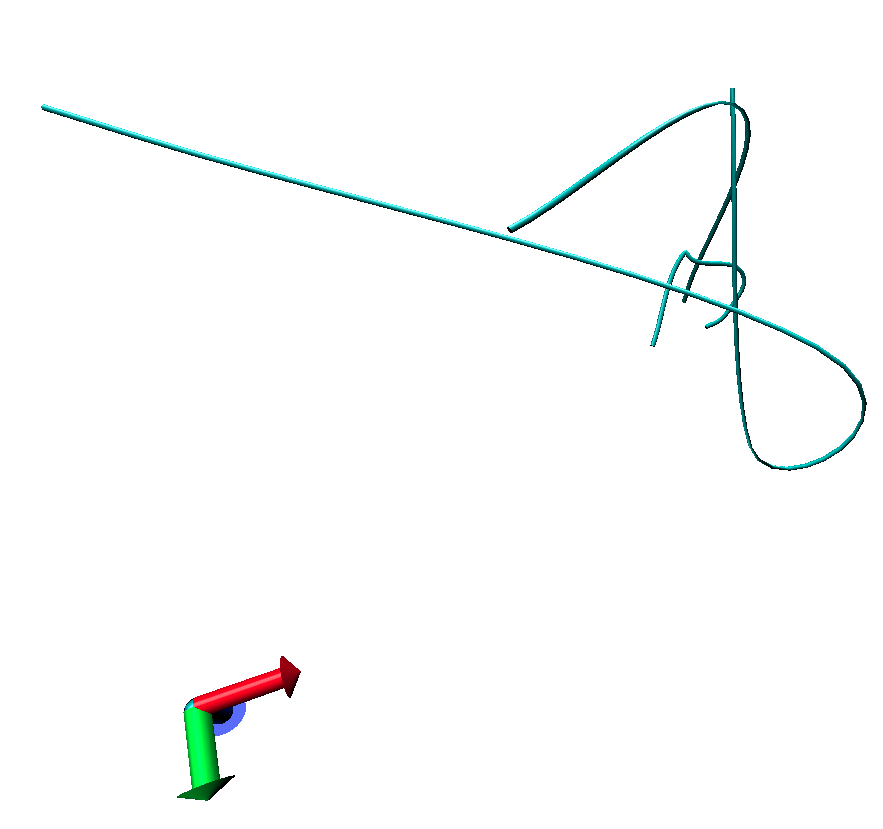
This results in a distorted peptide and water molecules. I have attached a
picture of the peptide below. I also tried with minimization and heating
steps using Amber14 and the result was the same in both TIP3P and MEOH
solvents.
The simulation runs fine and shows no error messages but the output is
distorted.
Also, I have used the same strategy for implicit simulation carrying out
minimization on AmberTools16 and production run on Amber14 and it turns out
to be fine always.
I have also checked running the production on AmberTools16 and this also
results in normal output.
I don't understand whether it is really the matter of using two different
versions of Amber or something else. I would be grateful if someone can
guide me towards the right direction.
Comparing the input files I don't find huge difference between AmberTools16
and Amber14
*Note: *I am trying to use Amber14 available on the local server. That is
the MPI version and is the latest one available.
[image: Inline image 3]
-- Best wishes Chetna
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
