Date: Tue, 14 Nov 2017 11:16:26 -0500
There was another typo in the method you provided us. You used
"saveamberpam" which should have been "saveamberparm".
Our point is that you need to tell us exactly what you did. Given the
typos, this is cannot be exactly what you did. If nothing else. without a
proper prmtop and inpcrd file, you could not have run a simulation.
So, we can only help you if you tell us exactly what you did.
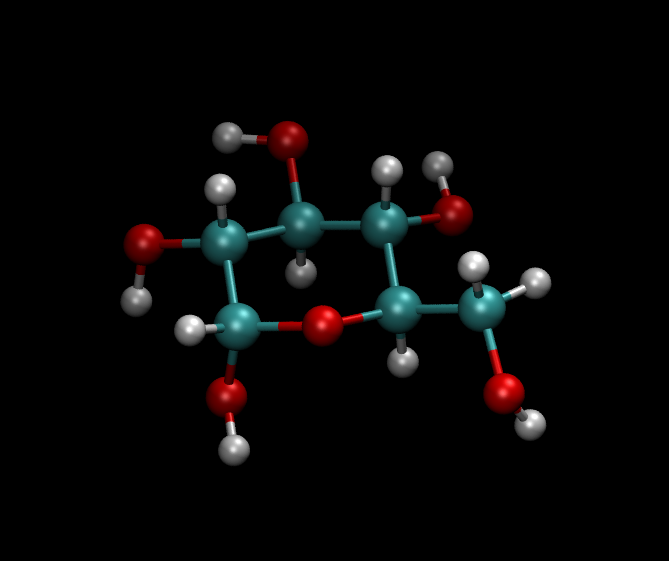
The following gave me a beautiful a-D-Glcp-1-OH (see image below). I
checked both the pdb file and the prmtop/impcrd combination using VMD. If
you got an apparent extra bond, it was something else that made it. Note
that an MD simulation will not create or destroy bonds. So, it is always
best to check structures using the prmtop/inpcrd file and not the pdb file.
I did this:
$ tleap -f leapin
Where:
$ cat leapin
source leaprc.GLYCAM_06j-1
GLC = sequence { ROH 0GA }
check GLC
saveoff GLC glc.lib
saveamberparm GLC glc.prmtop glc.inpcrd
savepdb GLC glc.pdb
quit
...and I got this image:
On Tue, Nov 14, 2017 at 10:27 AM, Aashish Bhatt <aashish.ph16221.inst.ac.in>
wrote:
> sorry sir is may mistake i have written check GLC not GLU.
>
> On Tue, Nov 14, 2017 at 8:53 PM, Carlos Simmerling <
> carlos.simmerling.gmail.com> wrote:
>
> > did you look at the leap.log file? For example, you wrote "check GLU" but
> > GLU was never defined. Leap must have told you there was a problem with
> > that. Did it give any other warnings or errors?
> >
> > On Tue, Nov 14, 2017 at 10:14 AM, Aashish Bhatt <
> > aashish.ph16221.inst.ac.in>
> > wrote:
> >
> > > I followed the step
> > >
> > > tleap -s -f leaprc.GLYCAM_06j-1
> > > GLC = sequence { ROH 0GA }
> > > check GLU
> > > saveoff GLC glc.lib
> > > saveamberpam GLC glc.prmtop glc.inpcrd
> > > savepdb GLC glc.pdb
> > > quit
> > >
> > >
> > >
> > > On Tue, Nov 14, 2017 at 8:33 PM, Carlos Simmerling <
> > > carlos.simmerling.gmail.com> wrote:
> > >
> > > > The existence of a bond isn't likely due to the charges. We don't
> know
> > > how
> > > > you set up all of the other parameters though, so it's hard to help.
> > > >
> > > > On Nov 14, 2017 9:48 AM, "Aashish Bhatt" <aashish.ph16221.inst.ac.in
> >
> > > > wrote:
> > > >
> > > > > Dear Sir/Ma'am
> > > > >
> > > > > I have performed glucose dynamics in vacuum using parameter
> > > > > (Mulliken,Lowedin,ESP,RESP and 0GA+ROH charges) that i got from
> orca
> > > and
> > > > > Gaussian dft optimization of glucose molecule. In gas phase
> dynamics
> > i
> > > > have
> > > > > found that the anomeric OH group is showing bonding to O(5) atom.
> > > > > I am not able to understand the reason behind this binding. I have
> > also
> > > > > attached screenshot of glucose molecule.
> > > > >
> > > > > Regards
> > > > > Aashish Bhatt
> > > > > PhD Scholar
> > > > > INST, Mohali
> > > > > India
> > > > >
> > > > > _______________________________________________
> > > > > AMBER mailing list
> > > > > AMBER.ambermd.org
> > > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > > >
> > > > >
> > > > _______________________________________________
> > > > AMBER mailing list
> > > > AMBER.ambermd.org
> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > >
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- :-) Lachele Lachele Foley CCRC/UGA Athens, GA USA
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: vmdscene.dat.png)
