Date: Thu, 12 Oct 2017 14:30:28 +0530
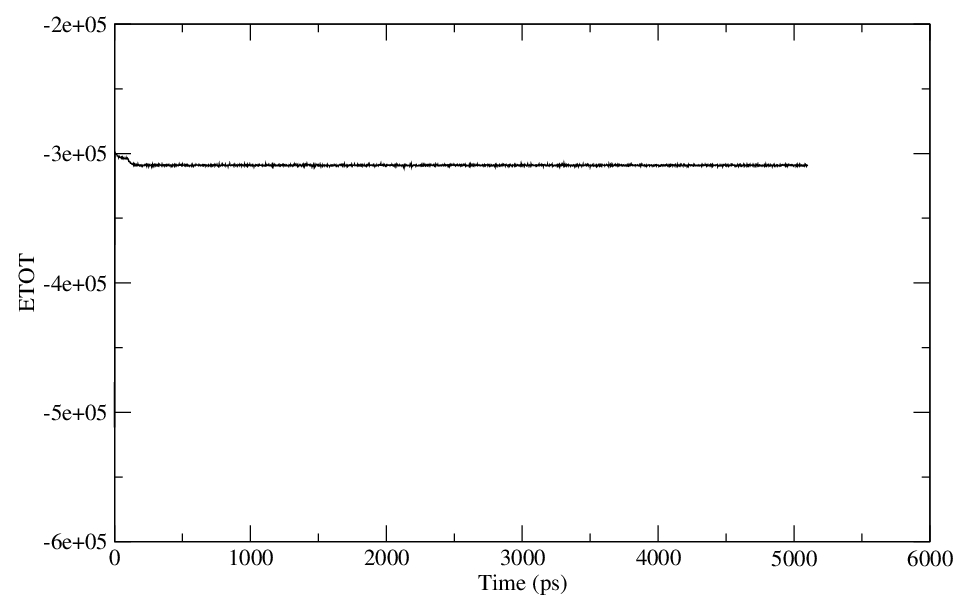
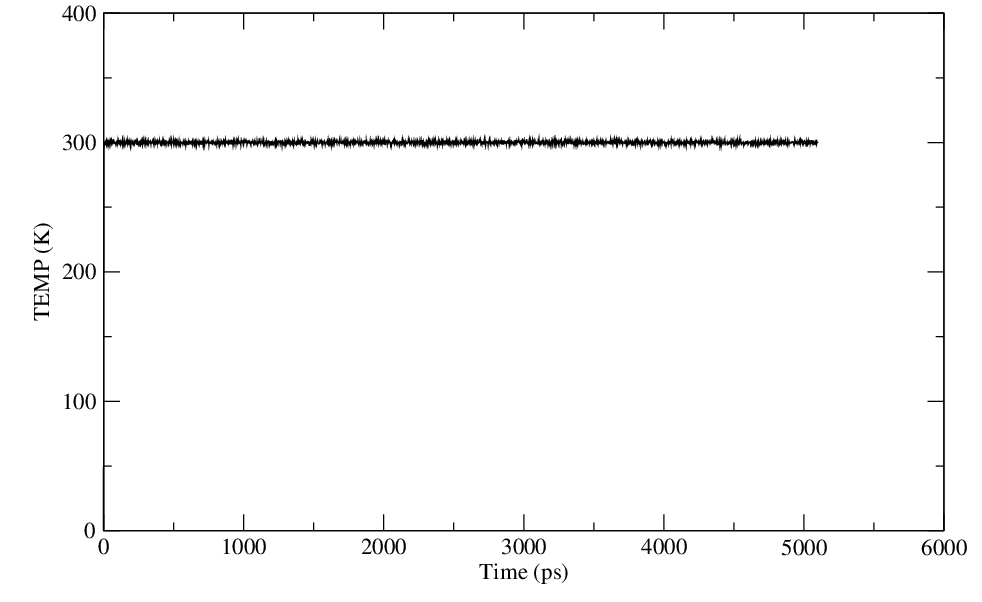
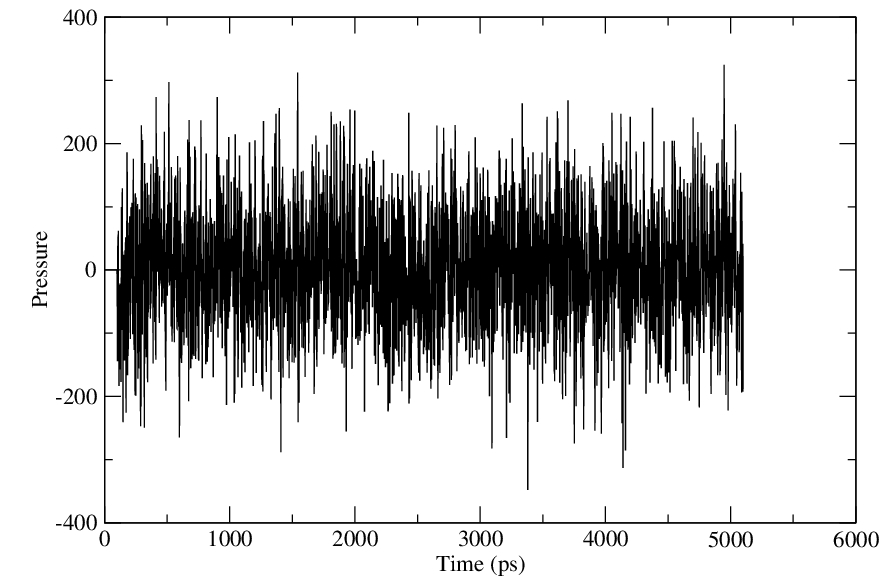
Yes, I have plotted and the energy, temperature have equilibrated properly.
On Thu, Oct 12, 2017 at 9:57 AM, Bill Ross <ross.cgl.ucsf.edu> wrote:
> Have you plotted and assessed them? Any conclusions?
>
>
> On 10/11/17 9:24 PM, Leena Aggarwal wrote:
> > I am attaching the files of energy, temperature and pressure.
> >
> > On Wed, Oct 11, 2017 at 11:01 PM, Bill Ross <ross.cgl.ucsf.edu> wrote:
> >
> >> Have you plotted energy, temperature etc yet?
> >>
> >>
> >> On 10/11/17 12:11 AM, Leena Aggarwal wrote:
> >>> There were no ************ at all in mdout , mdcrd and mdrst before
> ~95
> >> ns.
> >>> ********* come in only the restart file after 95 ns.
> >>>
> >>> On Wed, Oct 11, 2017 at 12:39 PM, Leena Aggarwal <leena.hrc.gmail.com>
> >>> wrote:
> >>>
> >>>> ************** comes in the output coordinate file after simulation
> run
> >> of
> >>>> ~95 ns
> >>>>
> >>>> On Wed, Oct 11, 2017 at 12:33 PM, Leena Aggarwal <leena.hrc.gmail.com
> >
> >>>> wrote:
> >>>>
> >>>>> I have not plotted but there are no ************ in the output
> >> coordinate
> >>>>> file.
> >>>>>
> >>>>> On Wed, Oct 11, 2017 at 11:35 AM, Bill Ross <ross.cgl.ucsf.edu>
> wrote:
> >>>>>
> >>>>>> Have you plotted temperature, pressure, energies and the like, to
> see
> >>>>>> how well-equilibrated the system is?
> >>>>>>
> >>>>>> Are there any *****'s in the .out files?
> >>>>>>
> >>>>>>
> >>>>>> On 10/10/17 9:17 PM, Leena Aggarwal wrote:
> >>>>>>> I am attaching the input files of equilibration. The number of
> atoms
> >>>>>> in the
> >>>>>>> system is around 98055. I have done the heating from 0 to 300K for
> >> 100
> >>>>>> ps
> >>>>>>> in NVT ensemble using Langevin thermostat. Then I have equilibrated
> >> the
> >>>>>>> system for 5 ns before production.
> >>>>>>>
> >>>>>>> On Tue, Oct 10, 2017 at 8:35 PM, Bill Ross <ross.cgl.ucsf.edu>
> >> wrote:
> >>>>>>>> How long did you equilibrate before production? Best to send those
> >> .in
> >>>>>>>> files too.
> >>>>>>>>
> >>>>>>>> Bill
> >>>>>>>>
> >>>>>>>>
> >>>>>>>> On 10/10/17 6:47 AM, Leena Aggarwal wrote:
> >>>>>>>>> I am attaching the input protocol file or production run input
> >> file.
> >>>>>>>>> On Tue, Oct 10, 2017 at 6:39 PM, Elvis Martis <
> >>>>>> elvis.martis.bcp.edu.in>
> >>>>>>>>> wrote:
> >>>>>>>>>
> >>>>>>>>>> HI,
> >>>>>>>>>> Can you also send you input protocol file?
> >>>>>>>>>>
> >>>>>>>>>>
> >>>>>>>>>> Best Regards
> >>>>>>>>>> Elvis Martis
> >>>>>>>>>> Mumbai, INDIA.
> >>>>>>>>>>
> >>>>>>>>>> ________________________________________
> >>>>>>>>>> From: Leena Aggarwal <leena.hrc.gmail.com>
> >>>>>>>>>> Sent: 10 October 2017 18:10
> >>>>>>>>>> To: amber.ambermd.org
> >>>>>>>>>> Subject: [AMBER] query regarding the restart file of molecular
> >>>>>> dynamics
> >>>>>>>>>> simulation
> >>>>>>>>>>
> >>>>>>>>>> I am getting '********************' in the restart file. So,
> while
> >>>>>>>>>> resubmitting the simulation it is showing the error : "getting
> new
> >>>>>> box
> >>>>>>>> info
> >>>>>>>>>> from bottom of inpcrd
> >>>>>>>>>> INFO : old style inpcrd file read".
> >>>>>>>>>>
> >>>>>>>>>> "************" error in restart file.
> >>>>>>>>>> ************-392.8431438 141.4329232************-393.5314779
> >>>>>>>> 142.1394832
> >>>>>>>>>> ************-392.6209402 141.4041265 512.3461308 -57.5257819
> >>>>>> 296.9723761
> >>>>>>>>>> 559.7273926-176.5667776************
> >>>>>> 560.1020382-176.1418198*******
> >>>>>>>> *****
> >>>>>>>>>> 559.0035464-175.9871851************ 156.3070715
> 146.8340440
> >>>>>>>> 417.5014573
> >>>>>>>>>> 155.9575107 183.8644124-287.7859939************
> 128.6822810
> >>>>>>>> -44.4453453
> >>>>>>>>>> ************ 127.7639674 -44.0672594************ 128.8132441
> >>>>>> -44.7128400
> >>>>>>>>>> _______________________________________________
> >>>>>>>>>> AMBER mailing list
> >>>>>>>>>> AMBER.ambermd.org
> >>>>>>>>>> http://lists.ambermd.org/mailman/listinfo/amber
> >>>>>>>>>>
> >>>>>>>>>> _______________________________________________
> >>>>>>>>>> AMBER mailing list
> >>>>>>>>>> AMBER.ambermd.org
> >>>>>>>>>> http://lists.ambermd.org/mailman/listinfo/amber
> >>>>>>>>>>
> >>>>>>>>>>
> >>>>>>>>>>
> >>>>>>>>>> _______________________________________________
> >>>>>>>>>> AMBER mailing list
> >>>>>>>>>> AMBER.ambermd.org
> >>>>>>>>>> http://lists.ambermd.org/mailman/listinfo/amber
> >>>>>>>> _______________________________________________
> >>>>>>>> AMBER mailing list
> >>>>>>>> AMBER.ambermd.org
> >>>>>>>> http://lists.ambermd.org/mailman/listinfo/amber
> >>>>>>>>
> >>>>>>>>
> >>>>>>>>
> >>>>>>>> _______________________________________________
> >>>>>>>> AMBER mailing list
> >>>>>>>> AMBER.ambermd.org
> >>>>>>>> http://lists.ambermd.org/mailman/listinfo/amber
> >>>>>> _______________________________________________
> >>>>>> AMBER mailing list
> >>>>>> AMBER.ambermd.org
> >>>>>> http://lists.ambermd.org/mailman/listinfo/amber
> >>>>>>
> >>> _______________________________________________
> >>> AMBER mailing list
> >>> AMBER.ambermd.org
> >>> http://lists.ambermd.org/mailman/listinfo/amber
> >>
> >> _______________________________________________
> >> AMBER mailing list
> >> AMBER.ambermd.org
> >> http://lists.ambermd.org/mailman/listinfo/amber
> >>
> >>
> >>
> >> _______________________________________________
> >> AMBER mailing list
> >> AMBER.ambermd.org
> >> http://lists.ambermd.org/mailman/listinfo/amber
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: total-energy.jpg)
(image/jpeg attachment: temp.jpg)
(image/jpeg attachment: pressure.jpg)
