Date: Mon, 28 Aug 2017 16:11:08 -0600
Dear David,
Thank you very much for your response. If I set the hybridization of n1 as
sp2 as it is in (leaprc.gaff) file, there is no problem but sp2 is not what
I want. I just don’t understand why there is discrepancy between the
hybridization of n1 in leaprc.gaff (sp2) and gaff.dat (sp) file. in
leaprc.gaff file all the nitrogens are sp2.
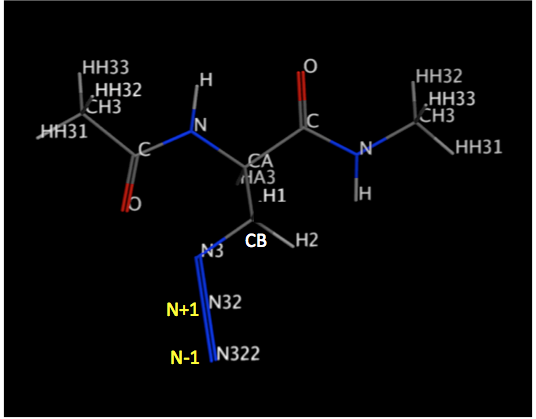
As I mentioned before, I tried to build a non-standard residue (Azide group
attached to ALA residue, I named it AZA) using pyRED and ANTECHAMBER.
Please check below for the pdb, mol2, leaprc.final and the frcmod that I
used for this purpose. I want the sp nitrogen since the angle between
Carbon (SP2) and Nitrogen (SP) attached to two other SP nitrogens should be
180 degrees. CB, N3, N32 and N322 should be in a line (please check the
attached figure).
I attached a figure of my non-standard residue to this email. The Azide
group contains three nitrogens that are attached to the carbon of ALA and
there is two double bond between three nitrogens so the second nitrogen
after the carbon of ALA has one extra electron (n1+) and the third one has
one electron less (n1-).
Here is the pdb file that I used in pyRED server:
ATOM 1 C1 ACE 0 -2.444 -2.719 -0.324
C
ATOM 2 C2 ACE 0 -2.429 -1.189 -0.324
C
ATOM 3 O2 ACE 0 -3.483 -0.555 -0.324
O
ATOM 4 H11 ACE 0 -3.465 -3.102 -0.324
H
ATOM 5 H12 ACE 0 -1.923 -3.072 -1.214
H
ATOM 6 H13 ACE 0 -1.923 -3.072 0.566
H
ATOM 7 N AZA 1 -1.251 -0.609 -0.324
N
ATOM 8 CA AZA 1 -1.117 0.833 -0.324
C
ATOM 9 CB AZA 1 -1.743 1.370 -1.489
C
*ATOM 10 N3 AZA 1 -1.629 2.793 -1.478
NATOM 11 N32 AZA 1 -1.514 4.216 -1.467
N1+ATOM 12 N322 AZA 1 -1.400 5.640 -1.456
N1-*
ATOM 13 C AZA 1 0.352 1.230 -0.324
C
ATOM 14 O AZA 1 1.230 0.370 -0.324
O
ATOM 15 H AZA 1 -0.419 -1.182 -0.324
H
ATOM 16 HA AZA 1 -1.595 1.243 0.566
H
ATOM 17 HB1 AZA 1 -2.796 1.090 -1.498
H
ATOM 18 HB2 AZA 1 -1.255 0.973 -2.379
H
ATOM 19 N1 NME 2 0.625 2.534 -0.324
N
ATOM 20 C3 NME 2 1.981 3.041 -0.324
C
ATOM 21 H1 NME 2 -0.141 3.192 -0.324
H
ATOM 22 H31 NME 2 1.962 4.132 -0.324
H
ATOM 23 H32 NME 2 2.502 2.688 0.566
H
ATOM 24 H33 NME 2 2.502 2.688 -1.214
H
*************************************************************
This is the mol2 file that I get fm pyRED server after charge calculation
using Gaussian .I choose the atomtypes using a hybrid approach from
(leaprc.gaff and leaprc.ff14SB). I change the N atoms related to the double
bond to the n1 atom as gaff.dat parameter symbol (in lower case). n1 in
gaff.dat is defined as: n1 14.01 0.530 Sp N since I
thought it is the best match hybridization wise.
.<TRIPOS>MOLECULE
AZA
12 11 1 0 1
SMALL
USER_CHARGES
.<TRIPOS>ATOM
1 N 1.333352 -0.884615 0.219577 N 1 AZA -0.4034 0.0000 ****
2 H 1.267148 -1.860126 0.413690 H 1 AZA 0.2693 0.0000 ****
3 CA 0.085939 -0.227084 -0.074450 CT 1 AZA -0.0432 0.0000 ****
4 HA 0.129415 0.209740 -1.066508 H1 1 AZA 0.1076 0.0000 ****
5 CB -0.173845 0.908895 0.934532 CT 1 AZA -0.0320 0.0000 ****
6 HB1 0.747648 1.457824 1.063615 H1 1 AZA 0.1195 0.0000 ****
7 HB2 -0.465700 0.497107 1.891496 H1 1 AZA 0.1195 0.0000 ****
8 N3 -1.264561 1.795275 0.495409 n1 1 AZA -0.5958 0.0000 ****
9 N32 -0.872516 2.853261 -0.004539 n1 1 AZA 0.6955 0.0000 ****
10 N322 -0.615656 3.824778 -0.449719 n1 1 AZA -0.3046 0.0000 ****
11 C -1.011458 -1.299221 -0.063064 C 1 AZA 0.6301 0.0000 ****
12 O -0.743596 -2.456283 0.143264 O 1 AZA -0.5625 0.0000 ****
.<TRIPOS>BOND
1 1 2 1
2 1 3 1
3 3 4 1
4 3 5 1
5 3 11 1
6 5 6 1
7 5 7 1
8 5 8 1
9 8 9 1
10 9 10 1
11 11 12 1
.<TRIPOS>SUBSTRUCTURE
1 AZA 1 **** 0 **** ****
.<TRIPOS>HEADTAIL
N 1
C 1
.<TRIPOS>RESIDUECONNECT
1 N C 0 0 0 0
*************************************************************
And get AZA.frcmod as below from ANTECHAMBER
parmchk -i AZA.mol2 -f mol3 -o AZA.frcmod
remark goes here
MASS
N 14.010 0.530 same as n
H 1.008 0.161 same as hn
CT 12.010 0.878 same as c3
H1 1.008 0.135 same as hc
C 12.010 0.616 same as c
O 16.000 0.434 same as o
BOND
N -H 410.20 1.009 same as hn-n
N -CT 330.60 1.460 same as c3-n
CT-H1 337.30 1.092 same as c3-hc
CT-CT 303.10 1.535 same as c3-c3
CT-C 328.30 1.508 same as c -c3
CT-n1 325.10 1.470 same as c3-n1
C -O 648.00 1.214 same as c -o
ANGLE
N -CT-H1 49.780 109.500 same as hc-c3-n
N -CT-CT 65.850 112.130 same as c3-c3-n
N -CT-C 66.670 111.560 same as c -c3-n
H -N -CT 46.040 116.780 same as c3-n -hn
CT-CT-H1 46.370 110.050 same as c3-c3-hc
CT-CT-n1 66.640 108.860 same as c3-c3-n1
CT-C -O 68.030 123.110 same as c3-c -o
H1-CT-C 47.200 109.680 same as c -c3-hc
CT-CT-C 63.790 110.530 same as c -c3-c3
CT-n1-n1 56.300 180.000 same as c3-n1-n1
H1-CT-H1 39.430 108.350 same as hc-c3-hc
H1-CT-n1 50.134 106.710 Calculated with empirical approach
DIHE
N -CT-CT-H1 1 0.156 0.000 3.000 same as X
-c3-c3-X
N -CT-CT-n1 1 0.156 0.000 3.000 same as X
-c3-c3-X
N -CT-C -O 1 0.000 180.000 2.000 same as X -c
-c3-X
H -N -CT-H1 1 0.000 0.000 2.000 same as X -c3-n
-X
H -N -CT-CT 1 0.000 0.000 2.000 same as X -c3-n
-X
H -N -CT-C 1 0.000 0.000 2.000 same as X -c3-n
-X
CT-CT-n1-n1 1 0.000 180.000 2.000 same as X
-n1-c3-X
H1-CT-CT-H1 1 0.150 0.000 3.000 same as
hc-c3-c3-hc
H1-CT-CT-n1 1 0.156 0.000 3.000 same as X
-c3-c3-X
H1-CT-C -O 1 0.800 0.000 -1.000 same as hc-c3-c
-o
H1-CT-C -O 1 0.080 180.000 3.000 same as hc-c3-c
-o
CT-CT-C -O 1 0.000 180.000 2.000 same as X -c
-c3-X
H1-CT-CT-C 1 0.156 0.000 3.000 same as X
-c3-c3-X
H1-CT-n1-n1 1 0.000 180.000 2.000 same as X
-n1-c3-X
n1-CT-CT-C 1 0.156 0.000 3.000 same as X
-c3-c3-X
IMPROPER
NONBON
N 1.8240 0.1700 same as n
H 0.6000 0.0157 same as hn
CT 1.9080 0.1094 same as c3
H1 1.4870 0.0157 same as hc
C 1.9080 0.0860 same as c
O 1.6612 0.2100 same as o
*************************************************************
leaprc.final file that I wrote as:
source leaprc.ff14SB
source leaprc.gaff
logfile q4md-forcefieldtools.log
# Web site: http://ambermd.org/doc6/html/AMBER-sh-5.9.html#sh-5.9.8
verbosity 2
# Web site: http://ambermd.org/doc6/html/AMBER-sh-5.9.html#sh-5.9.2
addAtomTypes {
{ "C" "C" "sp2" }
{ "CT" "C" "sp3" }
{ "H" "H" "sp3" }
{ "H1" "H" "sp3" }
{ "HC" "H" "sp3" }
{ "n1" "N" "sp” }
{ "n1" "N" "sp” }
{ "n1" "N" "sp” }
{ "O" "O" "sp2" }
}
frcmod1 = loadAmberParams AZA.frcmod
AZA = loadmol2 AZA.mol2
set AZA name "AZA"
set AZA head AZA.1.N
set AZA tail AZA.1.C
set AZA.1 connect0 AZA.1.N
set AZA.1 connect1 AZA.1.C
set AZA.1 restype protein
set AZA.1 name "AZA"
set AZA.1.N type N
set AZA.1.H type H
set AZA.1.CA <http://aza.1.ca/> type CT
set AZA.1.HA type H1
set AZA.1.CB type CT
set AZA.1.HB1 type H1
set AZA.1.HB2 type H1
set AZA.1.N3 type n1
set AZA.1.N32 type n1
set AZA.1.N322 type n1
set AZA.1.C type "C"
set AZA.1.O type O
saveoff AZA AZA.lib
SaveAmberParm AZA AZA.top AZA.crd
*************************************************************
This is what I get when I use { "n1" "N" "sp” } in Addatomtype
session.
atom type n1 - unknown hybridization sp
If I use { "n1" "N" "sp2” } instead there is no problem.
why there is discrepancy between the hybridization of n1 in leaprc.gaff
(sp2) and gaff.dat (sp) file?
Best regards,
Julian
----------------------------------------------
Julian Stys, Undergrad in Physics and Computer Science
Co-op Student
Department of Chemical and Materials Engineering
University of Alberta
On Sun, Aug 27, 2017 at 3:08 PM, David A Case <david.case.rutgers.edu>
wrote:
> On Fri, Aug 25, 2017, Julian Stys wrote:
> >
> > I am making a non-standard residue (Azide group attached to ALA residue),
> > and I'm using missing parameters from both gaff.dat and parm99.dat. For
> > nitrogens, I'm using (n1 14.01 0.530 Sp N), and so I added addAtomTypes{
> {
> > "n1" "N" "sp" } } in my leaprc script.
> >
> > The problem is that when I try to make the lib file, the hybridization
> > changes to sp2 because all the nitrogen types in leaprc.gaff are sp2. It
> > gives me the error: atom type n1 - unknown hybridization sp.
>
> First: are you sure this is an error message? There are three places in
> amber.c where the hybridization is checked, and only one of them allows
> "sp"
> as an option (no idea why right now). But it's also not clear from a
> quick look at the code that this message triggers any error.
>
> In general, LEaP makes very little use of "hybridizations". You could try
> setting n1 to sp2 in your leaprc file (as in leaprc.gaff) and see what
> happens. Unless LEaP has to try to build systems with missing
> coordinates, it
> doesn't look like type "sp" is every used.
>
> Let us know what happens: at a first glance it appears to be an oversight
> (bug) that hybridization "sp" is not being accepted.
>
> ...thanks for the report....dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screen_Shot_2017-08-28_at_3.47.22_PM.png)
