Date: Thu, 20 Jul 2017 02:39:39 +0530
Thanks for reply David.
I have used following command
For ion unbound protein
for i in 0 1 2 #3 4 5 6 7 8 9
do
j=$(( $i + 1 ))
/net/software/local/intel/impi/5.0.3.049/intel64/bin/mpirun
<http://s.bl-1.com/h/LYgodFG?url=http://5.0.3.049/intel64/bin/mpirun> -np
24 /net/software/local/amber/amber16/bin/pmemd.MPI -i
prod.in <http://s.bl-1.com/h/LYgokfJ?url=http://prod.in/> -o prod-$j.out -p
L_carb-protein.prmtop -c prod-$i.rst -r prod-$j.rst -x prod-$j.mdcrd
For ion bound protein
for i in 0 1 2 #3 4 5 6 7 8 9
do
j=$(( $i + 1 ))
/net/software/local/intel/impi/5.0.3.049/intel64/bin/mpirun
<http://s.bl-1.com/h/LYgoo2L?url=http://5.0.3.049/intel64/bin/mpirun> -np
24 /net/software/local/amber/amber16/bin/pmemd.MPI -i
prod.in <http://s.bl-1.com/h/LYgosRN?url=http://prod.in/> -o prod-$j.out -p
L_carb_solv_1264.prmtop -c prod-$i.rst -r prod-$j.rst -x prod-$j.mdcrd
These commands show that I am not using wrong restart file.
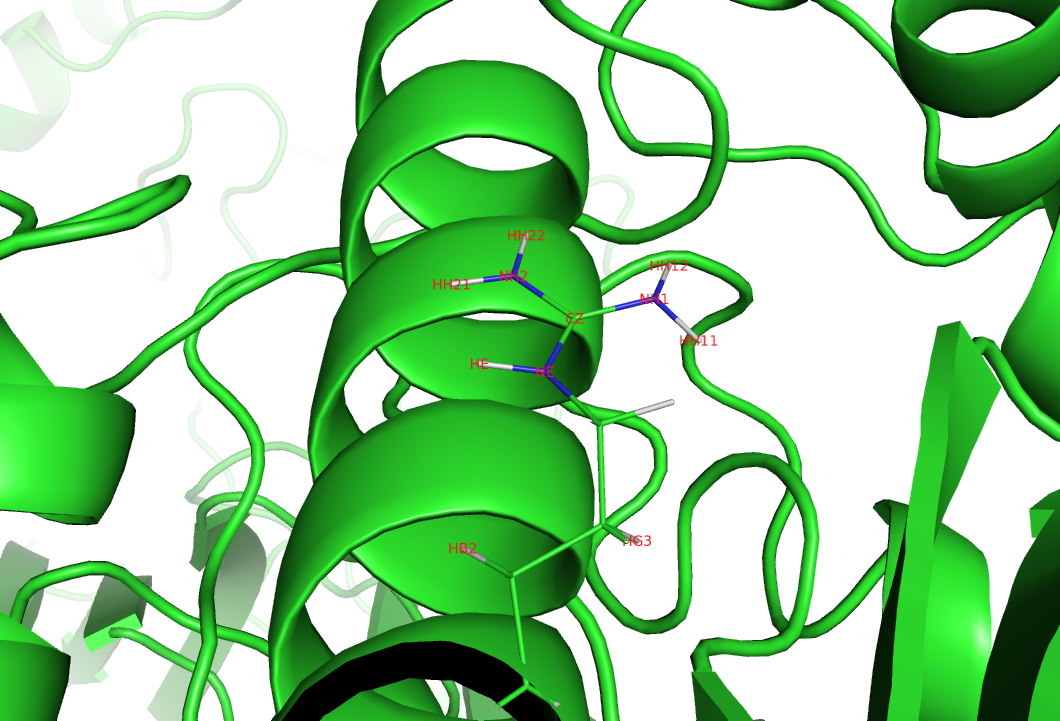
I also checked geometry of atoms 6893 and 6894 in the pdb file generated
from .rst file of first 5ns production run. Geometry of both atoms seems
ok. Herewith I am attaching .png file showing 6893 (NH2) and 6894(HH21)
atoms.
I am getting this error on one computer but when using another computer,
there is no such error, so could it be possible that this error is a bug?
Thanks
On Wed, Jul 19, 2017 at 9:21 PM, David A Case <david.case.rutgers.edu>
wrote:
> On Wed, Jul 19, 2017, sangita kachhap wrote:
> >
> > I am studying metalloprotein using MD simulation and for this, I have
> used
> > metal ion modeling tutorial of AMBER.
> > I am doing minimization of metal ion bound protein in two steps-1st with
> > restraint and 2nd without restraint, heating at NVT with restraint,
> > releasing restraint in three steps, equilibration at NPT and the
> production
> > run at NVT. Till production run of 5 ns, everything is fine but when
> > restarting production after 5 ns getting following error.
> >
> > vlimit exceeded for step 0; vmax = **********
> >
> > Coordinate resetting cannot be accomplished,
> > deviation is too large
> > iter_cnt, my_bond_idx, i and j are : 2 22 6893 6894
>
> This sounds like something is wrong with the restart file. Double/triple
> check that you didn't just have a wrong file name, or that you are really
> restarting from the exact file you finished with in the early step in the
> run
>
> You can create a PDB file from the restart file, and look at the geometry
> around atoms 6893 and 6894, which presumably are far from their idea
> geometry.
>
> It doesn't look like this has anything to do with the presence or absence
> of metal ions.
>
> ....dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: atom_6893-6894.png)
