Date: Thu, 1 Jun 2017 19:50:28 +0300
Dear David,
thank you very much for your explanations.
Would like to ask you and other Amber users
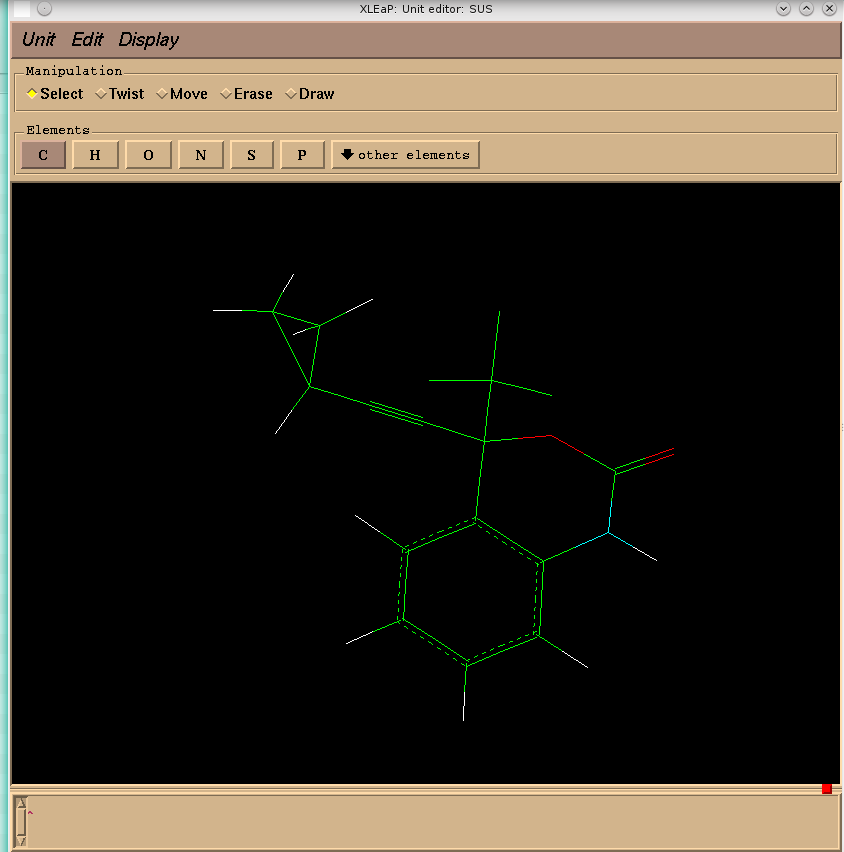
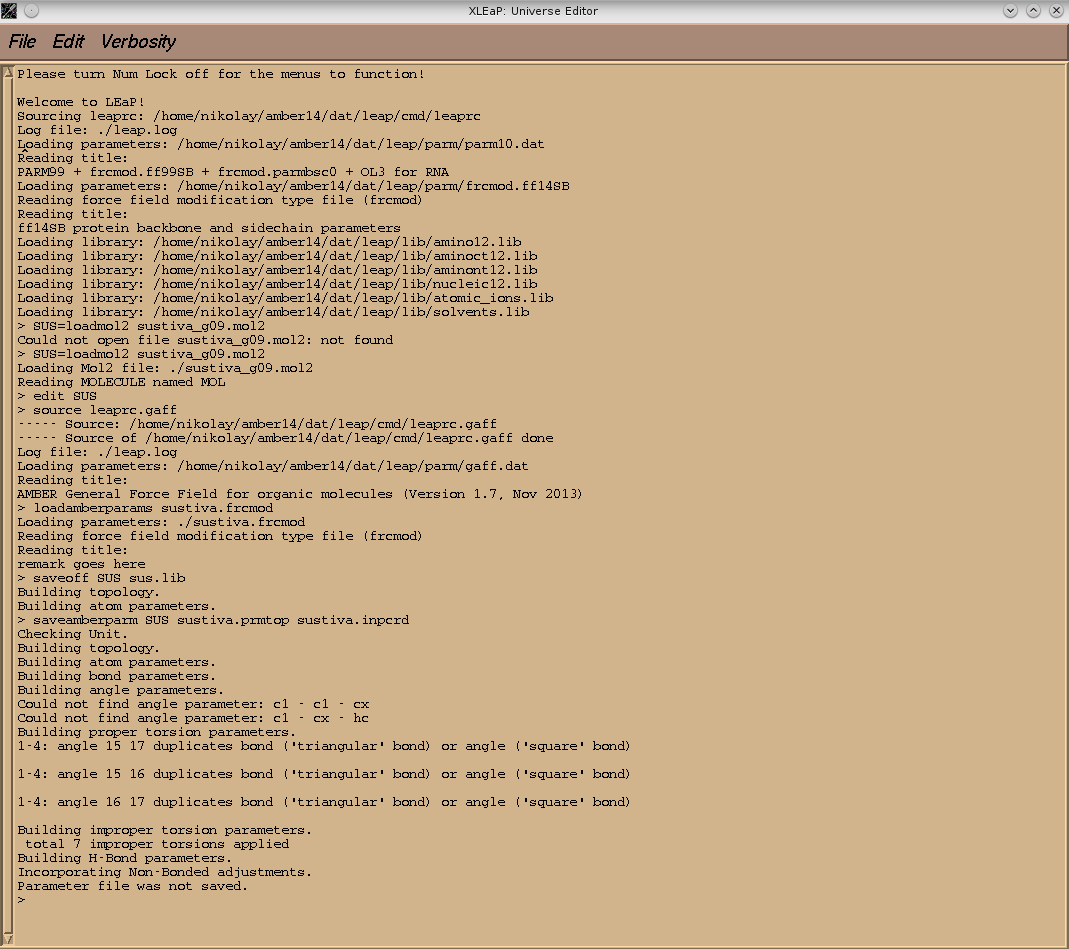
why xleap refused to create prmtop and inpcrd files for sustiva molecule
(there is a screenshot from xleap window enclosed).
It was not OK with the parameters in the tutorial example either,
but in my case the discrepancies turned out to be more critical.
Although the Gaussian-optimized structure looked nice...
Thank you in advance,
Nick
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Sustiva_xleap2.png)
(image/png attachment: Sustiva_xleap.png)
