Date: Mon, 17 Apr 2017 15:38:55 +0000
Thank you so much.
I have got confused then.
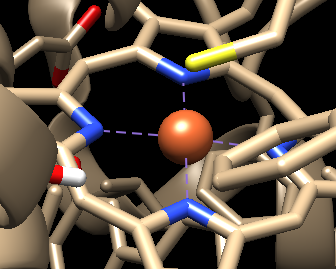
One more small question does it show any bond if I visualise it in chimera or anything? As after giving bond command I am unable to find any Hydrogen on Sulphur of CYS but chimera is not showing any bond.
Should I consider it to be bonded?
I am attaching screen shot for your reference.
________________________________
From: Adrian Roitberg <roitberg.ufl.edu>
Sent: Monday, April 17, 2017 4:14:58 AM
To: AMBER Mailing List
Subject: Re: [AMBER] coordinate bond between them and S and FE
Hi Thakur
I believe we are confusing things a bit.
You wrote before
"But bond command will form hydrogen bond, I want a coordinate bond"
I just asked where did you got that from, since the bond command CLEARLY makes what you call coordinate bonds, and I pointed you to the manual.
Adrian
On 4/17/17 10:45 AM, Thakur, Abhishek wrote:
> Hi Dr.Adian
>
>
> I am sure it a coordinate bond as it is between Fe of HEME and S of CYS. I am attaching a screen shot of my system for your reference
>
>
> Will this bond command make coordinate bonds?
>
>
> With regards,
>
> -AT
>
>
> ________________________________
> From: Adrian Roitberg <roitberg.ufl.edu>
> Sent: Friday, April 14, 2017 9:48:37 AM
> To: AMBER Mailing List
> Subject: Re: [AMBER] coordinate bond between them and S and FE
>
> why do you say that ?
>
>
> From the amber16 manual:
>
>
> 13.5.9. bond
>
> bond atom1 atom2 [ order ]
>
> Create a bond between atom1 and atom2. Both of these ATOMs must be
> contained by the same UNIT. By
> default, the bond will be a single bond. By specifying “-”, “=”, “#”, or
> “:” as the optional argument, order, the
> user can specify a single, double, triple, or aromatic bond,
> respectively. Example:
>
> bond trx.32.SG trx.35.SG
>
>
>
>
> On 4/14/17 4:44 PM, Thakur, Abhishek wrote:
>> But bond command will form hydrogen bond, I want a coordinate bond
>>
>> Sent using OWA for iPhone
>> ________________________________
>> From: Marcos Serrou do Amaral <marcossamaral.gmail.com>
>> Sent: Friday, April 14, 2017 8:24:54 AM
>> To: AMBER Mailing List
>> Subject: Re: [AMBER] coordinate bond between them and S and FE
>>
>> On Fri, Apr 14, 2017 at 3:18 PM, Thakur, Abhishek <axt651.miami.edu> wrote:
>>
>>> Can you suggest me how to tell tleap to make it a coordinate bond. I am
>>> afraid as by giving a bond command it will form hydrogen bond with it.
>>>
>> You need to use 'bond' command between S and a carbon atom. If your S atom
>> has a hydrogen atom, you need to throw it away. I am not sure, but I think
>> it is possible to do it in leap.
>>
>> Good luck!
>> ---
>> Marcos S.A.
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> https://urldefense.proofpoint.com/v2/url?u=http-3A__lists.ambermd.org_mailman_listinfo_amber&d=DwICAg&c=y2w-uYmhgFWijp_IQN0DhA&r=0Hah93XWYYBgmROOVcaccg&m=ff1IFhq4n_Mz0QT21AGb0WJAx5ziLlmA5nMBXNcElBQ&s=I6y-5f1Ik2PCxFr18GfWKV4XsxG56-O7L9Ws3x6ZJKU&e=
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> https://urldefense.proofpoint.com/v2/url?u=http-3A__lists.ambermd.org_mailman_listinfo_amber&d=DwIF-g&c=y2w-uYmhgFWijp_IQN0DhA&r=0Hah93XWYYBgmROOVcaccg&m=ckH95NwElHBzemH8aPZMzJhXX48S2MB1YpmvuRwCJ-c&s=i5jl3DhLMyLhFtMkd2GdAHJkt0ZvbW6R0ybdjPE8aUA&e=
> --
> Dr. Adrian E. Roitberg
> University of Florida Research Foundation Professor.
> Department of Chemistry
> University of Florida
> roitberg.ufl.edu
> 352-392-6972
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> https://urldefense.proofpoint.com/v2/url?u=http-3A__lists.ambermd.org_mailman_listinfo_amber&d=DwIF-g&c=y2w-uYmhgFWijp_IQN0DhA&r=0Hah93XWYYBgmROOVcaccg&m=ckH95NwElHBzemH8aPZMzJhXX48S2MB1YpmvuRwCJ-c&s=i5jl3DhLMyLhFtMkd2GdAHJkt0ZvbW6R0ybdjPE8aUA&e=
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> https://urldefense.proofpoint.com/v2/url?u=http-3A__lists.ambermd.org_mailman_listinfo_amber&d=DwIF-g&c=y2w-uYmhgFWijp_IQN0DhA&r=0Hah93XWYYBgmROOVcaccg&m=FXv9jc3FWdu3EvgdczDyu3kKkruC8HrLUpAXmPnTGQw&s=-nv8vY031UYtC66hY41grN6WW3wG2tzCb1m2Cu_rgzE&e=
-- Dr. Adrian E. Roitberg University of Florida Research Foundation Professor Department of Chemistry University of Florida roitberg.ufl.edu 352-392-6972 _______________________________________________ AMBER mailing list AMBER.ambermd.org https://urldefense.proofpoint.com/v2/url?u=http-3A__lists.ambermd.org_mailman_listinfo_amber&d=DwIF-g&c=y2w-uYmhgFWijp_IQN0DhA&r=0Hah93XWYYBgmROOVcaccg&m=FXv9jc3FWdu3EvgdczDyu3kKkruC8HrLUpAXmPnTGQw&s=-nv8vY031UYtC66hY41grN6WW3wG2tzCb1m2Cu_rgzE&e=
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screen_Shot_2017-04-17_at_11.37.56_AM.png)
