Date: Sun, 9 Apr 2017 22:40:02 +0100
Thank you for your advice. How do you suggest I could resolve the vdw term?
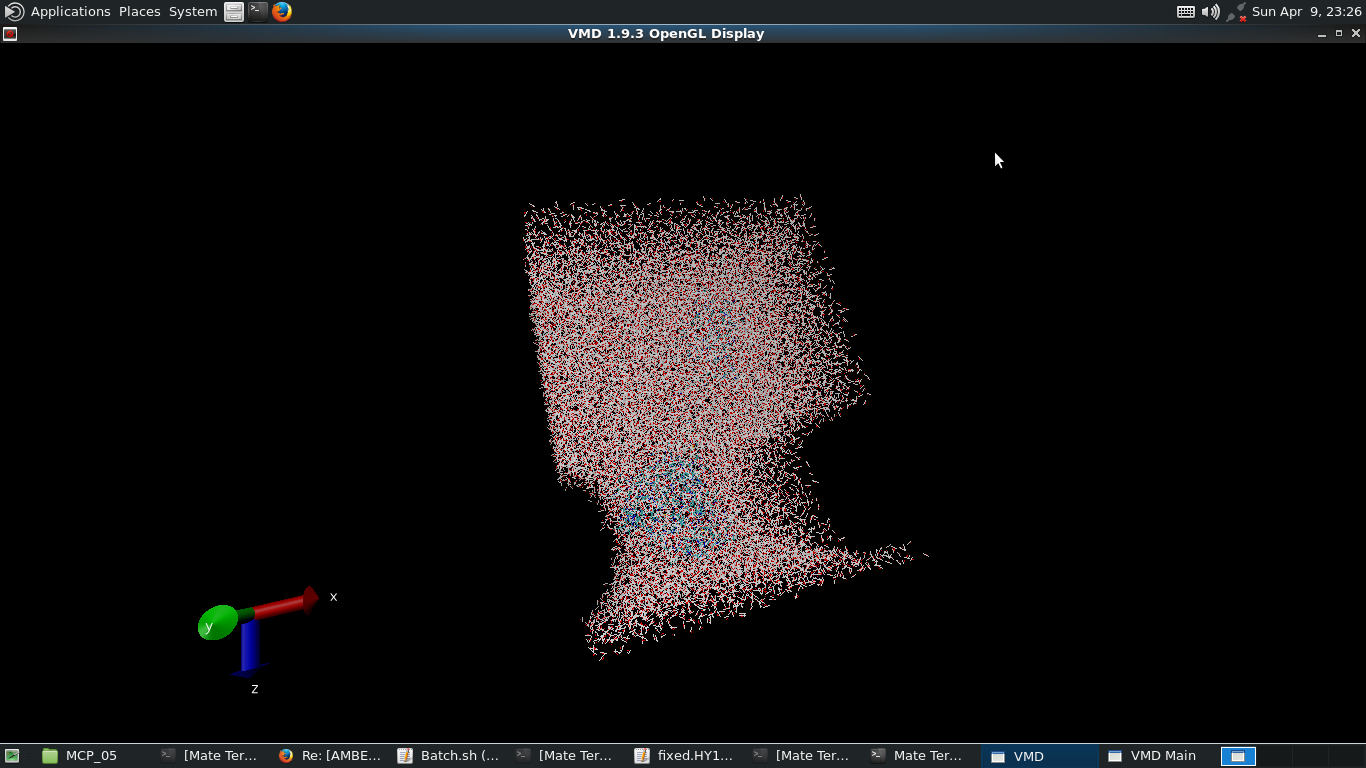
I am currently running some minimization and nvt relaxation with restraints on my protein, tautp=5, iwrap=1, and 1 fs time steps. Which leads to an oddly shaped water structure (attached). Not sure if this is due to the periodicity or not.
Von meinem iPad gesendet
> Am 09.04.2017 um 22:06 schrieb Chris Moth <cmoth08.gmail.com>:
>
> While he BOND energy is concerning, I'd resolve the van der Waals term first
>
> In my experience, when you see energies outside the display bounds *****.
>
> BOND = 258342.4685 ANGLE = 1028.6606 DIHED = 3160.9109
> VDWAALS = ************* <<<!!!
>
> on step 1, the next geometry is exploded far away from the start coordinates (along the few bad contacts) - and things go from bad to worse for step 2.
>
> Good luck.
>
> Chris
>
>
>
>
>> On 4/9/2017 12:43 AM, Hai Nguyen wrote:
>> On Sat, Apr 8, 2017 at 7:08 PM, Andreas Tosstorff <
>> andreas.tosstorff.cup.uni-muenchen.de> wrote:
>>
>>>> Hi all,
>>>>
>>>> I am having trouble with minimizing a structure I generated with MCPB.py.
>>>>
>>>> The minimization always fails after ncyc steps, apparently independently
>>> of what ncyc actually is and without any specific error message. I tried
>>> setting ntpr=1 but also here no specific error.
>>>> I run the minimization with pmemd and sander. See below for an excerpt
>>> of the output. I tried sending it with the complete file attached but the
>>> mail bounced back.
>>>>
>>
>>
>>>> I checked my structure with cpptraj check, and there are some close
>>> contacts, and a bond that is too long. However I was hoping to fix this by
>>> minimization. If required I can provide the topology and coordinates file.
>>>
>>
>> Hi,
>>
>> I think in this case, you need to see the WARNING from tleap about long
>> (usually incorrect) bond.
>> If the bond was created wrongly, don't mind about fixing by minimization at
>> all.
>>
>> Hai
>>
>>
>>>> I would really appreciate your help!
>>>>
>>>>
>>>> Best,
>>>>
>>>> Andy
>>> -------------------------------------------------------
>>> Amber 16 PMEMD 2016
>>> -------------------------------------------------------
>>>
>>> | PMEMD implementation of SANDER, Release 16
>>>
>>> | Run on 04/07/2017 at 12:53:51
>>>
>>> | Executable path: pmemd
>>> | Working directory: /state/partition2/home/local/
>>> andtos-loc/Dropbox/PhD/MCP/MCP_05
>>> | Hostname: kemi-steno.win.dtu.dk
>>>
>>> [-O]verwriting output
>>>
>>> File Assignments:
>>> | MDIN: min.in
>>> | MDOUT: fixed.HY133_solv_spc_min.out
>>> | INPCRD: fixed.restart.HY133_solv_spc.inpcrd
>>> | PARM: fixed.HY133_solv_spc.prmtop
>>> | RESTRT: fixed.HY133_solv_spc_min.rst
>>> | REFC: refc
>>> | MDVEL: mdvel
>>> | MDEN: mden
>>> | MDCRD: mdcrd
>>> | MDINFO: mdinfo
>>> | MDFRC: mdfrc
>>>
>>>
>>> Here is the input file:
>>>
>>>
>>> &cntrl
>>> imin=1, maxcyc=10000, ncyc=6000, ntpr=1,
>>> &end
>>>
>>>
>>> Note: ig = -1. Setting random seed to 409910 based on wallclock time in
>>> microseconds.
>>>
>>>
>>> | Conditional Compilation Defines Used:
>>> | PUBFFT
>>> | BINTRAJ
>>> | EMIL
>>>
>>> | Largest sphere to fit in unit cell has radius = 36.488
>>>
>>> | New format PARM file being parsed.
>>> | Version = 1.000 Date = 04/07/17 Time = 10:28:32
>>>
>>> | Note: 1-4 EEL scale factors are being read from the topology file.
>>>
>>> | Note: 1-4 VDW scale factors are being read from the topology file.
>>> | Duplicated 0 dihedrals
>>>
>>> | Duplicated 0 dihedrals
>>>
>>> ------------------------------------------------------------
>>> --------------------
>>> 1. RESOURCE USE:
>>> ------------------------------------------------------------
>>> --------------------
>>>
>>> getting new box info from bottom of inpcrd
>>> NATOM = 81317 NTYPES = 21 NBONH = 79101 MBONA = 2265
>>> NTHETH = 4859 MTHETA = 3097 NPHIH = 10115 MPHIA = 9652
>>> NHPARM = 0 NPARM = 0 NNB = 126684 NRES = 25954
>>> NBONA = 2265 NTHETA = 3097 NPHIA = 9652 NUMBND = 81
>>> NUMANG = 192 NPTRA = 191 NATYP = 46 NPHB = 1
>>> IFBOX = 1 NMXRS = 24 IFCAP = 0 NEXTRA = 0
>>> NCOPY = 0
>>>
>>> | Coordinate Index Table dimensions: 22 16 28
>>> | Direct force subcell size = 4.6501 4.5610 4.6290
>>>
>>> BOX TYPE: RECTILINEAR
>>>
>>> ------------------------------------------------------------
>>> --------------------
>>> 2. CONTROL DATA FOR THE RUN
>>> ------------------------------------------------------------
>>> --------------------
>>>
>>> default_name
>>>
>>> General flags:
>>> imin = 1, nmropt = 0
>>>
>>> Nature and format of input:
>>> ntx = 1, irest = 0, ntrx = 1
>>>
>>> Nature and format of output:
>>> ntxo = 2, ntpr = 1, ntrx = 1, ntwr =
>>> 1
>>> iwrap = 0, ntwx = 0, ntwv = 0, ntwe =
>>> 0
>>> ioutfm = 1, ntwprt = 0, idecomp = 0, rbornstat=
>>> 0
>>>
>>> Potential function:
>>> ntf = 1, ntb = 1, igb = 0, nsnb =
>>> 25
>>> ipol = 0, gbsa = 0, iesp = 0
>>> dielc = 1.00000, cut = 8.00000, intdiel = 1.00000
>>>
>>> Frozen or restrained atoms:
>>> ibelly = 0, ntr = 0
>>>
>>> Energy minimization:
>>> maxcyc = 10000, ncyc = 6000, ntmin = 1
>>> dx0 = 0.01000, drms = 0.00010
>>>
>>> | Intermolecular bonds treatment:
>>> | no_intermolecular_bonds = 1
>>>
>>> | Energy averages sample interval:
>>> | ene_avg_sampling = 1
>>>
>>> Ewald parameters:
>>> verbose = 0, ew_type = 0, nbflag = 1, use_pme =
>>> 1
>>> vdwmeth = 1, eedmeth = 1, netfrc = 0
>>> Box X = 102.302 Box Y = 72.976 Box Z = 129.612
>>> Alpha = 90.000 Beta = 90.000 Gamma = 90.000
>>> NFFT1 = 108 NFFT2 = 75 NFFT3 = 135
>>> Cutoff= 8.000 Tol =0.100E-04
>>> Ewald Coefficient = 0.34864
>>> Interpolation order = 4
>>>
>>> ------------------------------------------------------------
>>> --------------------
>>> 3. ATOMIC COORDINATES AND VELOCITIES
>>> ------------------------------------------------------------
>>> --------------------
>>>
>>> Cpptraj Generated Restart
>>> begin time read from input coords = 0.000 ps
>>>
>>>
>>> Number of triangulated 3-point waters found: 25653
>>>
>>> Sum of charges from parm topology file = -0.00002930
>>> Forcing neutrality...
>>>
>>> | Dynamic Memory, Types Used:
>>> | Reals 1914841
>>> | Integers 2157536
>>>
>>> | Nonbonded Pairs Initial Allocation: 13581971
>>>
>>> ------------------------------------------------------------
>>> --------------------
>>> 4. RESULTS
>>> ------------------------------------------------------------
>>> --------------------
>>>
>>> ---------------------------------------------------
>>> APPROXIMATING switch and d/dx switch using CUBIC SPLINE INTERPOLATION
>>> using 5000.0 points per unit in tabled values
>>> TESTING RELATIVE ERROR over r ranging from 0.0 to cutoff
>>> | CHECK switch(x): max rel err = 0.2738E-14 at 2.422500
>>> | CHECK d/dx switch(x): max rel err = 0.8332E-11 at 2.782960
>>> ---------------------------------------------------
>>> |---------------------------------------------------
>>> | APPROXIMATING direct energy using CUBIC SPLINE INTERPOLATION
>>> | with 50.0 points per unit in tabled values
>>> | Relative Error Limit not exceeded for r .gt. 2.47
>>> | APPROXIMATING direct force using CUBIC SPLINE INTERPOLATION
>>> | with 50.0 points per unit in tabled values
>>> | Relative Error Limit not exceeded for r .gt. 2.89
>>> |---------------------------------------------------
>>>
>>>
>>> NSTEP ENERGY RMS GMAX NAME NUMBER
>>> 1 5.0426E+08 3.8287E+07 1.0554E+10 HA3 2568
>>>
>>> BOND = 258342.4685 ANGLE = 1028.6606 DIHED =
>>> 3160.9109
>>> VDWAALS = ************* EEL = -284621.7195 HBOND =
>>> 0.0000
>>> 1-4 VDW = 1565.2178 1-4 EEL = 13661.6160 RESTRAINT =
>>> 0.0000
>>>
>>>> --
>>>> M.Sc. Andreas Tosstorff
>>>> Lehrstuhl für Pharmazeutische Technologie und Biopharmazie
>>>> Department Pharmazie
>>>> LMU München
>>>> Butenandtstr. 5-13 ( Haus B)
>>>> 81377 München
>>>> Germany
>>>> Tel.: +49 89 2180 77059
>>> _______________________________________________
>>> AMBER mailing list
>>> AMBER.ambermd.org
>>> http://lists.ambermd.org/mailman/listinfo/amber
>>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_at_2017-04-09_23-26-37.png)
