Date: Sat, 25 Feb 2017 02:32:11 +0000
Dear all,
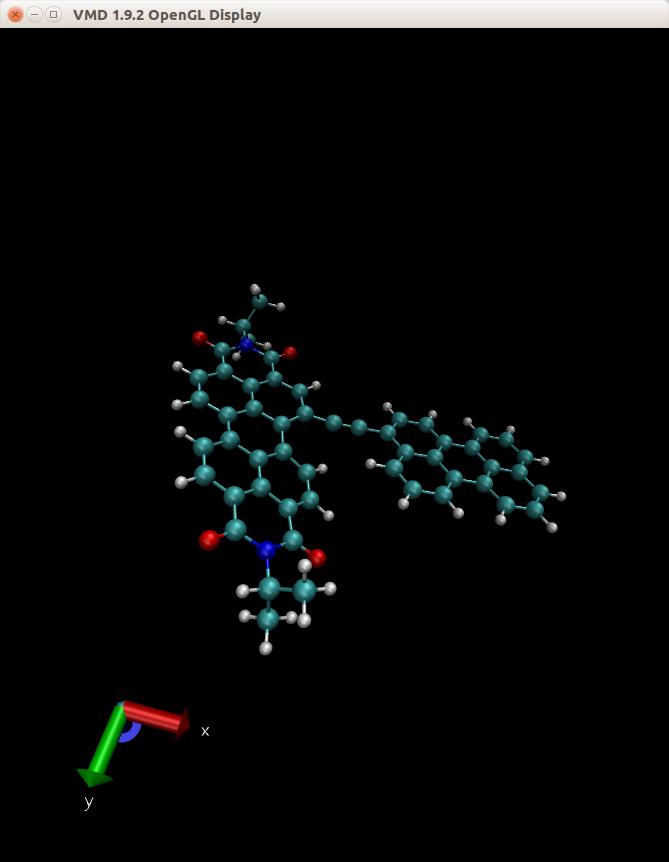
I'm trying to run simulations for molecules containing structures such as
C6H5-C-triple-bond-C-C6H5
using GAFF2 force field. (I copy the Hai Long's No answer Question)(In my case is PDI-C-triple bond-C-PE)
Due to the electron interaction between these two fragments (Benzene-Benzen or my case PDI-PE),
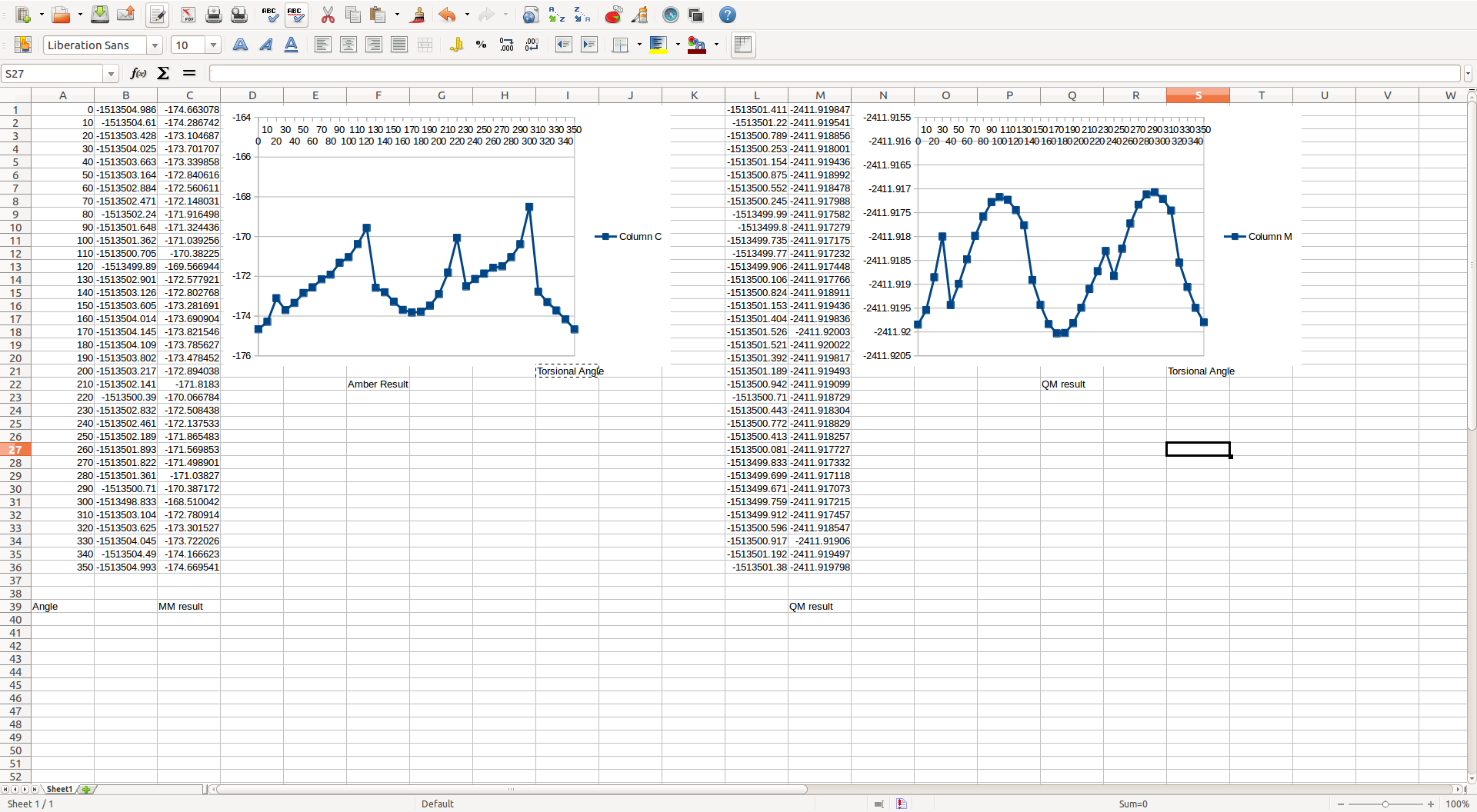
when the two fragments have different torsional angle, the energy of the molecule will change.
But due to the damning C-triple bond-C linking, it is very difficult to define the Dihedral angle which I plan to use "parmfit" to make Amber energy equal to QM energy result!
So I checked gaff force field for sp carbon parameters. I know all of the dihedral parameters for sp carbon such as cg(or ch)
(Inner Sp carbons in conjugated systems) have zero PK value. For example:
X -cg-cg- X 1 0.000 180.000 2.000
X -cg-ch- X 1 0.000 180.000 2.000
for both triple and single bonds
This is because this line structure can not define a Dihedral angle mathematically!
But We(Hai Long's 2011 Question and me) need a way to parmfit our molecule's "Dihedral" potential crossing this C-triple-bond-C.
Please help me to this question,
and my molecule and potential energy curve of amber and QM are attached.
Many Thanks,
Siwei Wang
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2017-02-24_20:41:51.png)
(image/png attachment: Screenshot_from_2017-02-24_20:42:18.png)
