Date: Tue, 13 Dec 2016 23:20:41 +0530
Thanks David.
I read the manual. Followed the tutorial 20 of metal-proteins MD once also.
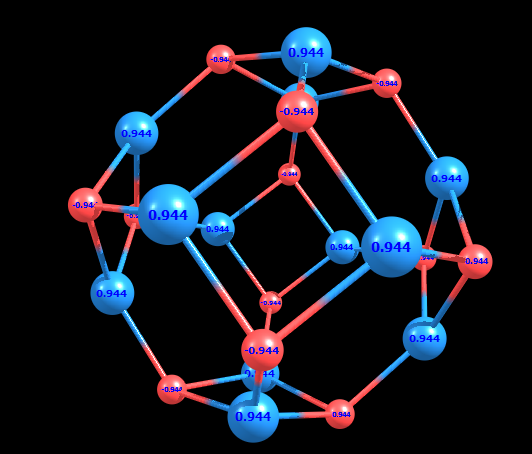
I am doubtful as my cluster is a cage like structure.
I am attaching a image to make things clear.
Please advice.
On Tue, Dec 13, 2016 at 9:04 PM, David A Case <david.case.rutgers.edu>
wrote:
> On Tue, Dec 13, 2016, vivek m wrote:
> >
> > I want to perform molecular dynamics of (ZnO)12 cluster with a
> protein.
> > I have drawn the ZnO cluster and performed gaussian optimization to get
> the
> > correct geometry and charges. I have also performed docking of ZnO
> cluster
> > with my protein. I have also done the protein preparation as described in
> > the AMBER tutorials.
> > I do not know how to move ahead from here. How do i prepare the ZnO-
> > protein prmtop and inpcrd files.
>
> See sections 15.7 and 15.8 in the Amber 2016 Reference Manual.
>
> ...dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: ZnO_cluster.png)
