Date: Mon, 28 Nov 2016 20:49:09 +0000
Dear AMBER users,
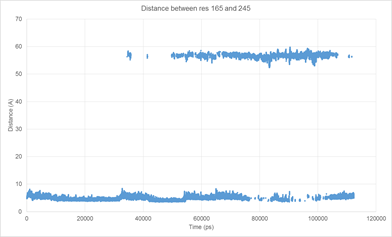
I'm hoping you can help me with imaging my system. My system consists of two interacting proteins - including repeats and variations of my proteins I have about 18 trajectory files. Normally, the two proteins end up separating from each other, with one of the interacting partners moving into a different box. I have had a large amount of success using autoimage in cpptraj to image 16/18 of them, but the remaining two are giving me a lot of problems. The problem seems to be the "ligand" protein jumping out of the periodic box. I've attached an example of what's happening using a bond distance between the interface of the two proteins. It might also be useful to note that my system won't RMS fit because the proteins move around the box.
[cid:image001.png.01D244A8.66C24D50]
My usual script would look something like this:
Parm x.top
Trajin simulation.x
Autoimage
Strip :WAT
Strip :NA
Trajout file.x
I've tried several variations on this to try and solve the problem, e.g.
Autoimage :1-172 or :173-260 (which are each protein respectively). I've also included the anchor command before specifying the mask as well with no luck.
I've also tried the following commands in different orders to see if that would solve the problem - still with no luck.
Autoimage
Center :1-260 mass origin (or :1-172/:173-260 as above)
Image origin center familiar
I've exhausted all of the previous solutions to problems on the reflector and don't know what to try next. Any ideas?
Many thanks,
Mark Waterhouse [RPG]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image001.png)
