Date: Thu, 24 Nov 2016 15:58:16 +0100
I have tried whole process of parametrization and minimization with
standard GAFF atom types. First I have generated mol2 file from output
of Gaussian esp calculation with:
antechamber -i FGrph-5hex_esp-charges-ini.esp -fi gesp -o
FGrph-5hex.mol2 -fo mol2 -c resp -eq 0 -at gaff -rn FGR
Than I have checked if all parameters for my system are defined in GAFF with:
parmchk -i FGrph-5hex.mol2 -f mol2 -o FGrph-5hex.frcmod
and there was no need to add any new parameters. Then I have created
parameter and coordinate file with tleap:
source leaprc.gaff
FGR=loadmol2 FGrph-5hex.mol2
loadamberparams FGrph-5hex.frcmod
saveamberparm FGR FGrph-5hex.prmtop FGrph-5hex.inpcrd
savepdb FGR FGrph-5hex.pdb
After that I have started minimization with sander:
sander -O -i 01_Min.in -o 01_Min.out -p FGrph-5hex.prmtop -c
FGrph-5hex.inpcrd -x FGrph-5hex.mdcrd -r 01_Min.rst -inf 01_Min.mdinfo
with input file 01_Min.in:
minimize
&cntrl
imin=1,
ntb=0,
ntx=1,
irest=0,
maxcyc=10000,
ncyc=4000,
ntpr=100,
ntwx=10,
cut=999.0,
/
and also nab calculation with input file:
molecule m;
float x[990], fret;
m = getpdb( "FGrph-5hex.pdb");
readparm( m, "FGrph-5hex.prmtop" );
mm_options( "cut=999.0, ntpr=50" );
setxyz_from_mol(m, NULL, x);
mme_init( m, NULL, "::ZZZ", x, NULL);
//conjugate gradient minimization
conjgrad(x, 3*m.natoms, fret, mme, 0.001, 0.0001, 30000);
//Newton-Raphson minimization
mm_options( "ntpr=10" );
newton( x, 3*m.natoms, fret, mme, mme2, 0.00000001, 0.0, 500 );
//Output minimized structure
setmol_from_xyz( m, NULL, x);
putpdb( "FGrph-5hex_min.pdb", m );
However, both structures (from sander minimization and nab
minimization) were again different (see the pictures in attachment). I
have no idea what I’m doing wrong. All files I have used are included
in the attachment.
Thank you for your help and suggestions.
>>
>> I have tried comparing energies and here are the results:
>> For initial configuration I receive from NAB:
>>
>> iter Total bad vdW elect nonpolar genBorn
>> frms
>> ff: 0 113013.79 4458.86 108611.29 -56.37 0.00 0.00
>> 5.38e+03
>>
>> and from sander:
>>
>> NSTEP ENERGY RMS GMAX NAME NUMBER
>> 1 4.9198E+03 1.7964E+01 7.5305E+01 C90 90
>>
>> BOND = 193.1278 ANGLE = 3623.6104 DIHED =
>> 642.1252
>> VDWAALS = 290.9881 EEL = 60.9590 HBOND =
>> 0.0000
>> 1-4 VDW = 226.0755 1-4 EEL = -117.0890 RESTRAINT =
>> 0.0000
>>
>> I think that there is something
>> wrong with my NAB input files or NAB reads bonds from prmtop file
>> somehow differently than sander.
>
> (I thought I had answered this earlier....) The results above show that the
> bond-angle-dihedral energy is identical in NAB and sander. So this almost
> certainly has nothing to do with how NAB reads bonds from the prmtop file.
>
> The difference is entirely in the vdW terms with the NAB energy being much
> higher than the sander result. Is there anything unusual about how you
> created the prmtop file? Have you tried a similar comparison using residues
> that are in the standard libraries? We have tests of exactly this sort of
> comparison in the Amber test suite, so there is something unusal about your
> specific situation. Any clues you can provide about anything "non-standard"
> that you did might help in debugging.
>
> ...dac
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- text/plain attachment: FGrph-5hex_esp-charges-ini.esp
- text/plain attachment: tleap.in
- text/x-c attachment: nmode.nab
- text/plain attachment: 01_Min.in
- application/x-sh attachment: run.sh
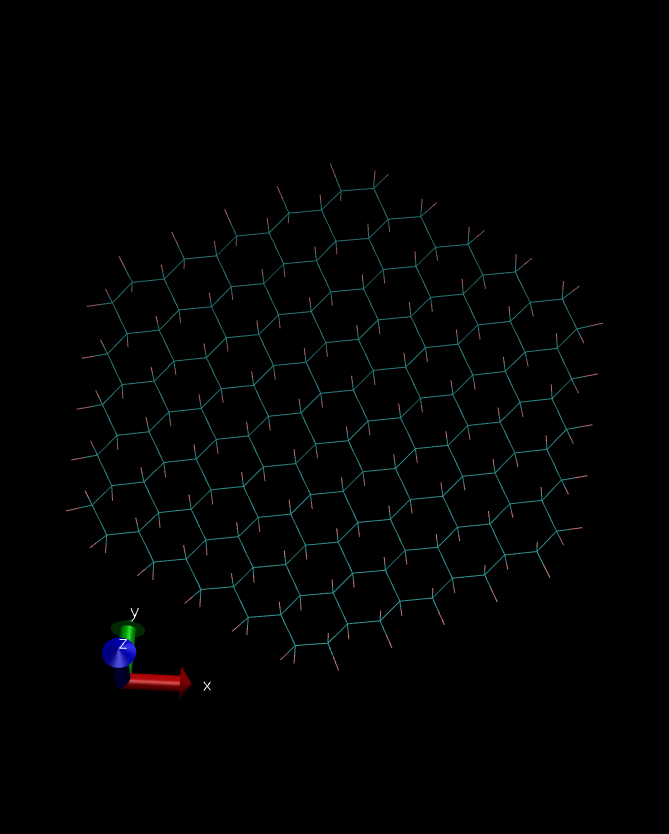
(image/png attachment: Sander-Min-Structure.png)
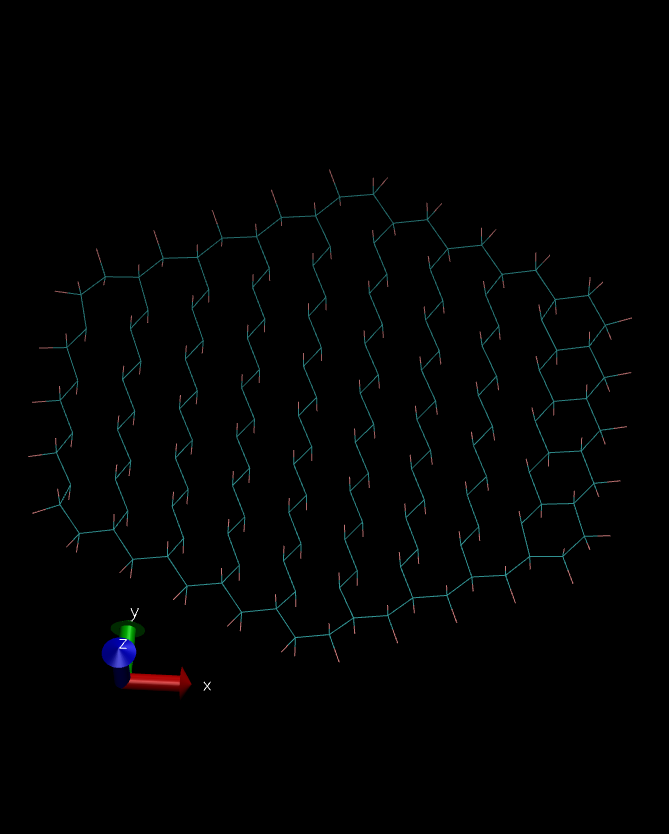
(image/png attachment: NAB-Min-Structure.png)
