Date: Thu, 10 Nov 2016 15:01:48 +0000
Dear all,
I have run into a curious problem when performing minimization with a FAD
co-factor, based on a Parmed-generated chamber prmtop.
Initially, a .psf file was generated with VMD's psfgen using CHARMM-style
parameters for FAD.
I confirmed that minimization with NAMD of FAD in vacuum, using the .psf
and .pdb file is ok.
(see min_namd.pdb attached).
Then I convert this psf and pdb with Parmed's chamber action (parmed
version 2.4.0 from AmberTools16):
chamber -top top_all36_prot.rtf -top top_all36_na.rtf -top
top_all36_cgenff.rtf -top jp4061522_si_002_mod.txt -param
par_all36_prot.prm -param par_all36_cgenff.prm -param par_all36_lipid.prm
-param jp4061522_si_003_mod.txt -str toppar_water_ions.str -psf
test_fad.psf -crd test_fad.pdb -box bounding
parmout test_fad.prmtop test_fad.rst
However, when I perform minimization with sander using the resulting
test_fad.prmtop and test_fad.rst files, something curious happens:
The central structure in the flavine ring distorts in a strange way - In
particular, C6A and C4A appear to exchange position, and both the
configuration around N5 and N10 is distorted.
(see min_test.pdb)
I've checked what parmed reports for the bonds of these atoms, and this is
all as expected.
Is this some strange bug in the writing of the prmtop?
I'm attaching an archive that contains the files required to run the above
Parmed-command.
Thank you in advance for looking into this!
Marc
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
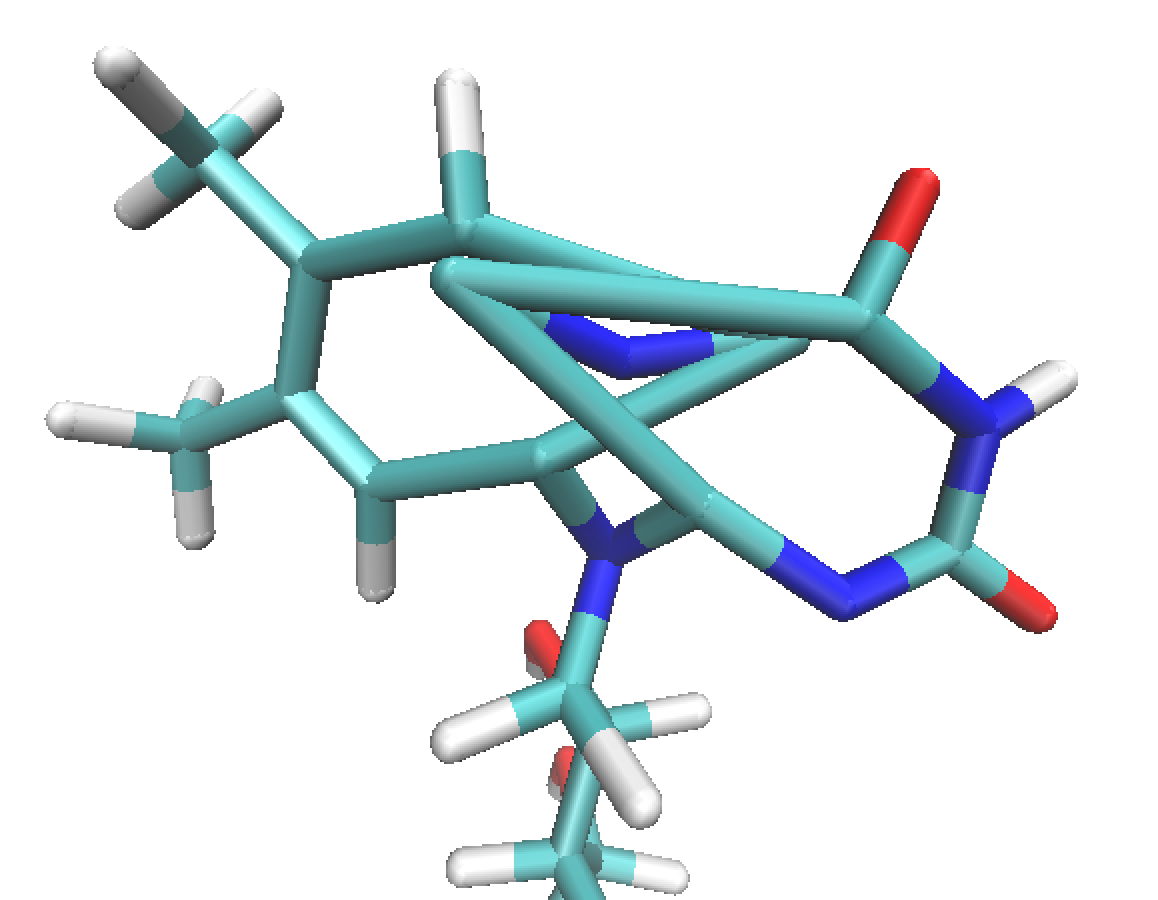
(image/png attachment: fad_distorted.png)
- application/x-gzip attachment: FAD-Parmed-Test.tar.gz
- chemical/x-pdb attachment: min_namd.pdb
- chemical/x-pdb attachment: min_test.pdb
