Date: Mon, 7 Nov 2016 08:38:25 +0100
Hi Fabricio and Callum,
Have u solved the nmr restraint problem? If so, would u help me and send
send me some input files as it may help me to create mine.
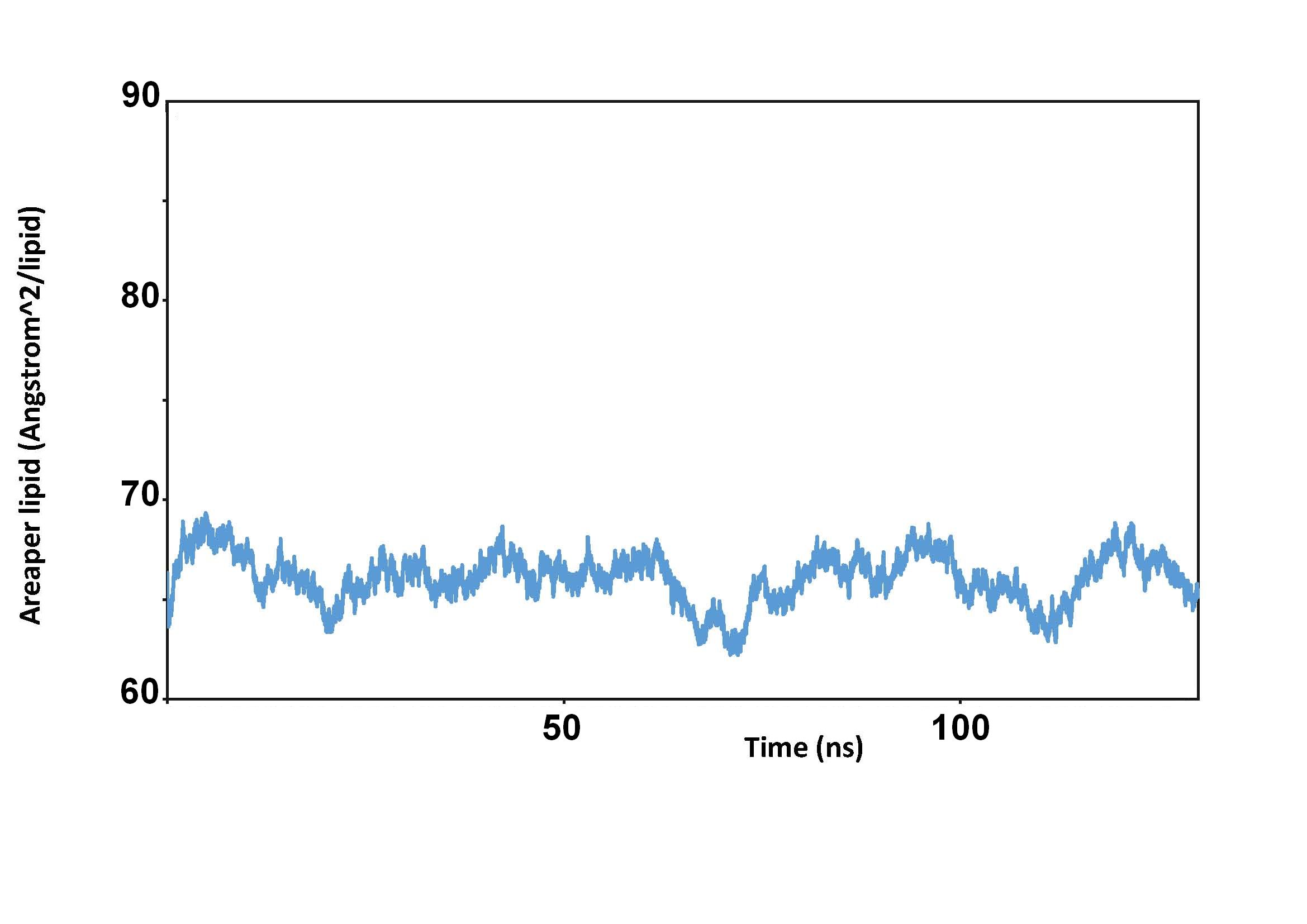
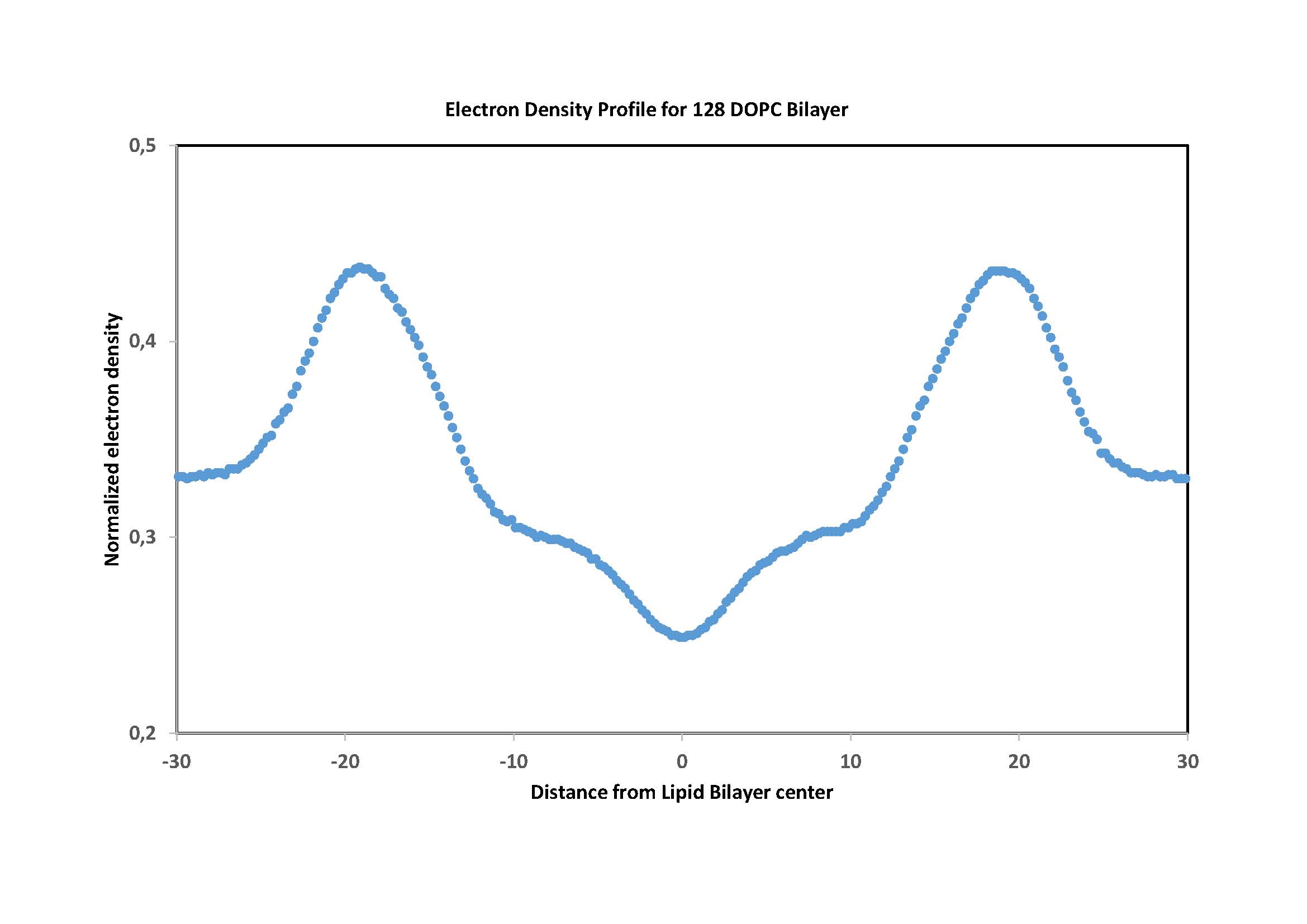
I simulated dynamics of system consisting of DOPC bilayer and my metal
(palladium) complexes. I obtained scholar results when Electron_density and
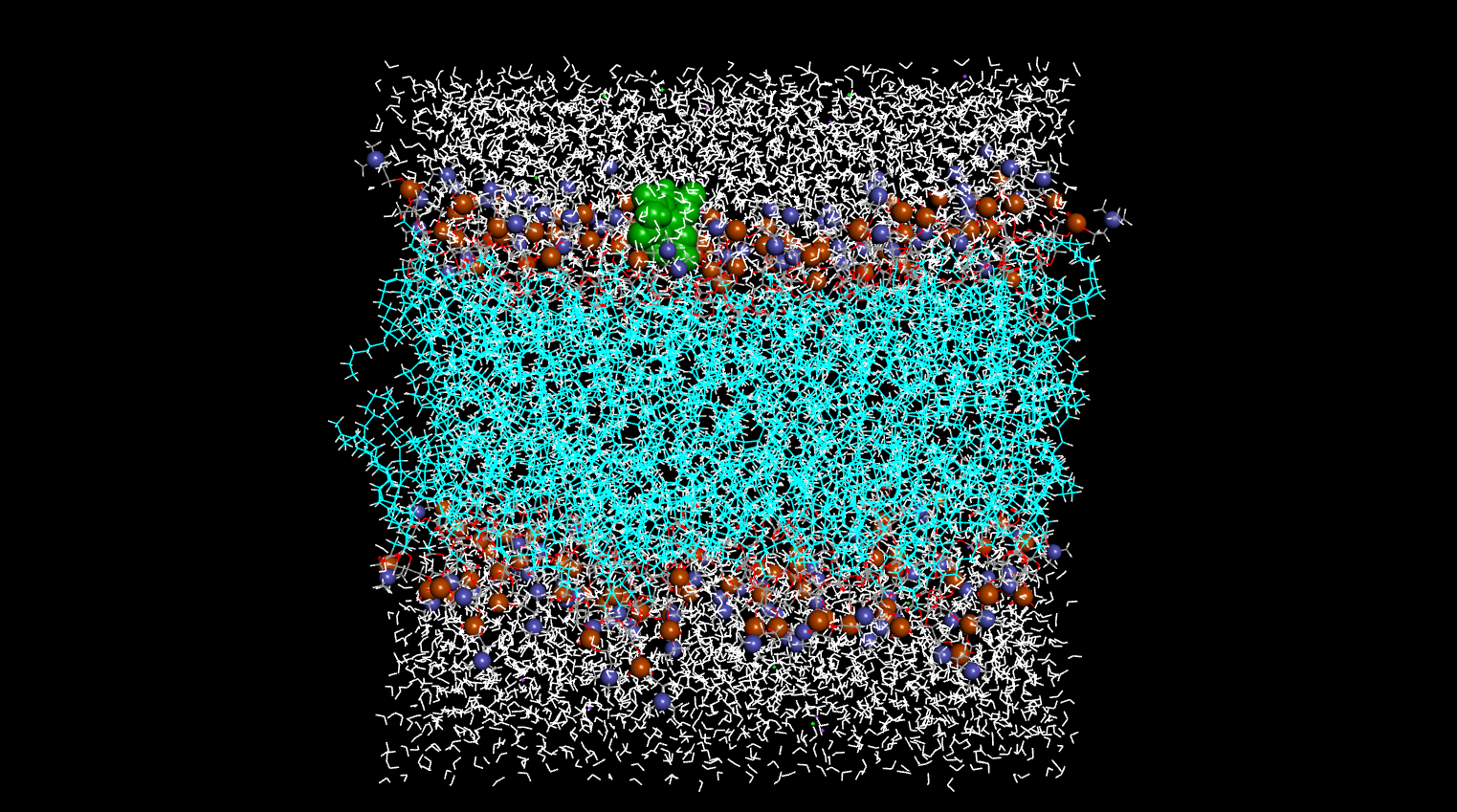
Area_per_lipid are in question (see pics). I also attached a pic describing
my system after 125ns of dynamics (attached one of the input production
files). What I want to know now is kind of distribution coefficient or
deltaG of diffusion of three different ligands (two my palladium complexes
and one cisplatin) through the lipid bilayer.
Would u people be so kind to help me (just in preparing input files or you
have some example input files on similar lipid-ligand systems) with this
regard? I can provide you with link for download of all my files (prmtop
or trajectory).
I would be very grateful if u take some time to help me.
Cheers
Zoran
-----Original Message-----
From: Fabrício Bracht
Sent: Thursday, October 13, 2016 4:40 PM
To: AMBER Mailing List
Subject: Re: [AMBER] Ligand across membrane
Hi Callum, the C16 and C17 are right in the middle of the bilayer. But I
will try with the nitrogen atoms.
I'll let you know if I have any problems.
Thanks
Fabrício
2016-10-12 10:52 GMT-03:00 Dickson, Callum J <
callum.dickson09.imperial.ac.uk>:
> Hi Fabricio,
>
>
> Where exactly are the C16 an C17 atoms in your lipids? I have used the
> fxyz functionality but using all nitrogen atoms in POPC lipid head groups
> (on both sides of the membrane, such that the center-of-mass defined by
> the
> nitrogens is approximately the middle of the bilayer). Or you could try
> the
> phosphorus atoms.
>
>
> Callum
>
> ________________________________
> From: Fabrício Bracht <fabracht1.gmail.com>
> Sent: Tuesday, October 11, 2016 8:26:15 PM
> To: AMBER Mailing List
> Subject: Re: [AMBER] Ligand across membrane
>
> Actually, I found the fxyz variable. The problem is that, for example, as
> I
> did, if you define the distance between the COM of the molecule and the
> lipid bilayer, you need to choose (max 1024, for pmemd, I think) atoms
> from
> the bilayer that will allow you change the distance, and that this
> distances will sample the crossing of the bilayer.
> I chose the c16 and c17 carbon atoms, but it did not work as expected. So,
> I was thinking if it would be possible to define a plane point distance or
> if the reaction coordinate can be defined relative to the box center.
> Or if you have any other suggestion, I would like to hear it.
> Thank you
> Fabrício
>
> Em 11 de out de 2016 6:14 PM, "Charles Lin" <clin92.ucsd.edu> escreveu:
>
> > I think you're looking for the fxyz nmr namelist variable (added in
> > Amber16)?
> >
> > Charlie
> > ________________________________________
> > From: Fabrício Bracht [fabracht1.gmail.com]
> > Sent: Tuesday, October 11, 2016 2:06 PM
> > To: AMBER Mailing List
> > Subject: [AMBER] Ligand across membrane
> >
> > Hi, I would like to know if there is a way to do umbrella sampling
> > simulations, using nmr restraints, of a ligand going across a lipid
> > bilayer. I have tried using COM pulling like so:
> >
> > &rst iat=-1,-1, iresid=0, r1=-100, r2=-4.5, r3=-4.5, r4=100.0, rk2 =
> > 40.,
> > rk3 = 40., fxyz=0,0,1,
> > igr1=30,152,314,317,362,446,570,573,618,702,786,910,913,
> > 920,1044,1047,1130,1254,1257,1342,1345,1430,1433,1518,1521,
> > 1528,1612,1736,1739,1824,1827,1912,1915,2000,2003,2088,2091,
> > 2176,2179,2186,2310,2313,2398,2401,2408,2608,2611,2618,2742,
> > 2745,2830,2833,2878,2962,3046,3130,3254,3257,3302,3426,3429,
> > 3704,3707,3792,3795,3878,4078,4081,4126,4250,4253,4300,4303,
> > 4310,4434,4437,4520,4644,4647,4732,4735,13142,13145,13230,
> > 13233,13240,13364,13367,13374,13458,13582,13585,13592,13754,
> > 13757,13764,13888,13891,13976,13979,14064,14067,14112,14196,
> > 14280,14364,14488,14491,14498,14582,14706,14709,14716,14800,
> > 14962,14965,14972,15172,15175,15260,15263,15270,15394,15397,
> > 15482,15485,15530,15654,15657,15742,15745,15752,15876,15879,
> > 15924,16048,16051,16058,16220,16223,16230,16352,16476,16479,
> > 16562,16646,16770,16773,16858,16861,16946,16949,17034,17037,
> > 17158,17320,17323,17330,17454,17457,17502,17702,17705,17712,
> > 17874,17877,17922,18046,18049,18134,18137,18184,18187,
> > igr2=1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19,
> 20,
> > 21, 22, 23, 24, 25, 26, 27, 28, 29, outxyz=1, /
> >
> > Where igr1 are all c16 atoms and c17 atoms of the lipids and igr2 is my
> > ligand. I used only c16 and c17 because of the atom limit in igr. But
> this
> > does not seem to work.
> > On a previous discussion (http://archive.ambermd.org/201602/0124.html)
> it
> > was mentioned something about the center of mass umbrella
> > restraint allowing the calculation of free energy of transfer profiles
> > to
> > move across a membrane bilayer.
> > Is that true, and, if so, how would one do that?
> >
> > Thanks
> > Fabrício
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: area_per_lipid.jpg)
(image/jpeg attachment: electron_density.jpg)
(image/png attachment: 4908.png)
- application/octet-stream attachment: 05_Prod.in
