Date: Wed, 5 Oct 2016 06:50:45 +0800
Dear sir,
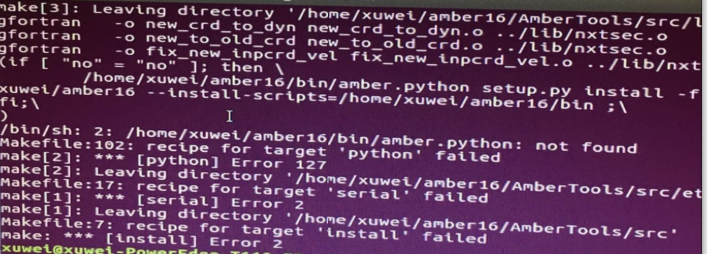
the /home/xuwei/amber16/bin directory exist. I tried to install the amber16 in ubuntu 16.04 operating system.
I attached the graph to make it clear.
From: amber-request
Date: 2016-10-05 03:00
To: amber
Subject: AMBER Digest, Vol 1717, Issue 1
Send AMBER mailing list submissions to
amber.ambermd.org
To subscribe or unsubscribe via the World Wide Web, visit
http://lists.ambermd.org/mailman/listinfo/amber
or, via email, send a message with subject or body 'help' to
amber-request.ambermd.org
You can reach the person managing the list at
amber-owner.ambermd.org
When replying, please edit your Subject line so it is more specific
than "Re: Contents of AMBER digest..."
AMBER Mailing List Digest
Today's Topics:
1. WHAM_ANALYSIS (Thakur, Abhishek)
2. missing windows qm/mm (Thakur, Abhishek)
3. Failing to compile AmberTools16 on Mac Sierra with Fink (Alan)
4. Re: Failing to compile AmberTools16 on Mac Sierra with Fink
(Jason Swails)
5. Re: MMPBSA.py error Your ligand residues must be sequential
in your complex (maryam azimzadehirani)
6. Re: MM-PBSA result for small molecule-receptor complex
(Maryam Hamzehee)
7. did not find amber.python (xwnail2003.163.com)
8. Re: did not find amber.python (Bill Ross)
9. Re: MM-PBSA result for small molecule-receptor complex
(David A Case)
10. time step changes during long simulation run (Hirdesh Kumar)
11. Re: time step changes during long simulation run (Jason Swails)
12. Re: time step changes during long simulation run (Hirdesh Kumar)
13. Re: MM-PBSA result for small molecule-receptor complex
(Bruno Falcone)
14. MD simulation by amber (Atila Petrosian)
----------------------------------------------------------------------
Message: 1
Date: Mon, 3 Oct 2016 19:49:46 +0000
From: "Thakur, Abhishek" <axt651.miami.edu>
Subject: [AMBER] WHAM_ANALYSIS
To: "amber.ambermd.org" <amber.ambermd.org>
Message-ID:
<CO2PR07MB266428E1074B616C55184AFA9EC20.CO2PR07MB2664.namprd07.prod.outlook.com>
Content-Type: text/plain; charset="iso-8859-1"
Hi Everyone,
I am facing little bit of problem in WHAM analysis.
In my system reaction coordinate start around 2.6 distance.
By after NPT the structure I have got has a distance of 3.9.
So in my WHAM analysis I have included the distance file from 3.9 and have got an energy barrier was 31k/cal and it seems intermediated was formed at 1.95 distance.
So as I have noticed that reaction started from 2.6 distance and getting some noise before that, so in my analysis this time I have included the output file from distance 2.7 only. But surprisingly I found that my energy barrier has reduced and intermediate has formed at 2.0 distance part.
Here below I have attached the the excel sheet and meta.dat files for both analysis.
Can anyone help me find why is this difference in result
Thanking you,
Abhishek
-------------- next part --------------
A non-text attachment was scrubbed...
Name: WHAM.xlsx
Type: application/vnd.openxmlformats-officedocument.spreadsheetml.sheet
Size: 47344 bytes
Desc: WHAM.xlsx
Url : http://lists.ambermd.org/mailman/private/amber/attachments/20161003/996ecbfb/attachment-0001.bin
-------------- next part --------------
A non-text attachment was scrubbed...
Name: meta_2.dat
Type: application/octet-stream
Size: 425 bytes
Desc: meta_2.dat
Url : http://lists.ambermd.org/mailman/private/amber/attachments/20161003/996ecbfb/attachment-0002.obj
-------------- next part --------------
A non-text attachment was scrubbed...
Name: meta.dat
Type: application/octet-stream
Size: 375 bytes
Desc: meta.dat
Url : http://lists.ambermd.org/mailman/private/amber/attachments/20161003/996ecbfb/attachment-0003.obj
------------------------------
Message: 2
Date: Mon, 3 Oct 2016 20:50:22 +0000
From: "Thakur, Abhishek" <axt651.miami.edu>
Subject: [AMBER] missing windows qm/mm
To: "amber.ambermd.org" <amber.ambermd.org>
Message-ID:
<CO2PR07MB266420EAE1DA124BD18245CF9EC20.CO2PR07MB2664.namprd07.prod.outlook.com>
Content-Type: text/plain; charset="iso-8859-1"
Hi everyone,
In my QM/MM calculation I can find that I am missing some windows in then can I just run those windows or I need to run from starting?
Thanking you,
AT
------------------------------
Message: 3
Date: Mon, 3 Oct 2016 22:26:26 +0100
From: Alan <alanwilter.gmail.com>
Subject: [AMBER] Failing to compile AmberTools16 on Mac Sierra with
Fink
To: AMBER Mailing List <amber.ambermd.org>
Message-ID:
<CAEznbzkN4DbXACxsiDFvz4J6moF4SjG3VJ+1w_NEaT-KeWurMw.mail.gmail.com>
Content-Type: text/plain; charset=UTF-8
Hi there,
I am wondering if someone else had succeeded, I am doing this:
- editting $AMBERHOME/AmberTools/src/configure2: use gcc-fsf-5 g++-fsf-5
gfortran-fsf-5
- cd $AMBERHOME; ./configure --with-python /sw/bin/python3.5 -macAccelerate
gnu
...
Compiling the NetCDF Fortran interface (may be time-consuming)...
Error: Could not compile with NetCDF Fortran interface.
gfortran-fsf-5 -fPIC -I/Users/alan/Programmes/amber16/include -o
testp testp.f90 /Users/alan/Programmes/amber16/lib/libnetcdff.a
/Users/alan/Programmes/amber16/lib/libnetcdf.a
Compile error follows:
Undefined symbols for architecture x86_64:
"_nf__create_", referenced from:
...
___netcdf_MOD_nf90_sync in libnetcdff.a(netcdf.o)
ld: symbol(s) not found for architecture x86_64
collect2: error: ld returned 1 exit status
Error: NetCDF build failed.
Configure failed due to the errors above!
Many thanks in advance,
Alan
--
Alan Wilter SOUSA da SILVA, DSc
Senior Bioinformatician, UniProt
European Bioinformatics Institute (EMBL-EBI)
European Molecular Biology Laboratory
Wellcome Trust Genome Campus
Hinxton
Cambridge CB10 1SD
United Kingdom
Tel: +44 (0)1223 494588
------------------------------
Message: 4
Date: Mon, 3 Oct 2016 19:32:12 -0400
From: Jason Swails <jason.swails.gmail.com>
Subject: Re: [AMBER] Failing to compile AmberTools16 on Mac Sierra
with Fink
To: AMBER Mailing List <amber.ambermd.org>
Message-ID:
<CAEk9e3rh8zhrobU3ErWbBAPn+FVV04xz5oxy0GZzYzjDsdKBNQ.mail.gmail.com>
Content-Type: text/plain; charset=UTF-8
On Mon, Oct 3, 2016 at 5:26 PM, Alan <alanwilter.gmail.com> wrote:
> Hi there,
>
> I am wondering if someone else had succeeded, I am doing this:
>
> - editting $AMBERHOME/AmberTools/src/configure2: use gcc-fsf-5 g++-fsf-5
> gfortran-fsf-5
> - cd $AMBERHOME; ./configure --with-python /sw/bin/python3.5 -macAccelerate
> gnu
>
> ...
> Compiling the NetCDF Fortran interface (may be time-consuming)...
> Error: Could not compile with NetCDF Fortran interface.
> gfortran-fsf-5 -fPIC -I/Users/alan/Programmes/amber16/include -o
> testp testp.f90 /Users/alan/Programmes/amber16/lib/libnetcdff.a
> /Users/alan/Programmes/amber16/lib/libnetcdf.a
> Compile error follows:
> Undefined symbols for architecture x86_64:
> "_nf__create_", referenced from:
> ...
> ___netcdf_MOD_nf90_sync in libnetcdff.a(netcdf.o)
> ld: symbol(s) not found for architecture x86_64
> collect2: error: ld returned 1 exit status
>
> Error: NetCDF build failed.
> Configure failed due to the errors above!
>
?For what it's worth, configure now respects CC, FC, and CXX environment
variables, so you no longer have to hack configure2 to use different GCC
compilers.
?Another idea is to build NetCDF using Fink and use "--with-netcdf" to
point to that NetCDF install. You can also use the "clang" compiler which
will use gfortran from Fink and clang/clang++ from XCode.
Just things to try. But I think MacPorts and Homebrew are more common
package managers used for Macs.
I've also tried just downloading a gfortran binary and using that with the
clang compilers and it's worked fine.
HTH,
Jason
--
Jason M. Swails
------------------------------
Message: 5
Date: Tue, 4 Oct 2016 11:11:51 +0800
From: maryam azimzadehirani <maryamai1988.gmail.com>
Subject: Re: [AMBER] MMPBSA.py error Your ligand residues must be
sequential in your complex
To: AMBER Mailing List <amber.ambermd.org>
Message-ID:
<CAEVOLfquqys93qarv6tq11XtoPPG2YqfYqZfZrK+GaquOdKnUA.mail.gmail.com>
Content-Type: text/plain; charset=UTF-8
Thank you Jason. You were right. It is working now.
On Mon, Oct 3, 2016 at 11:00 PM, Jason Swails <jason.swails.gmail.com>
wrote:
> On Sun, Oct 2, 2016 at 8:53 PM, maryam azimzadehirani <
> maryamai1988.gmail.com> wrote:
>
> > Hi,
> > I did try the mask and I am still getting the same error. To make sure I
> > ran a new short simulation and ran the analysis and I get this error:
> >
> > Loading and checking parameter files for compatibility...
> > mmpbsa_py_energy found! Using
> > /usr/local/packages/amber12/amber12/bin/mmpbsa_py_energy
> > cpptraj found! Using /usr/local/packages/amber12/amber12/bin/cpptraj
> >
>
> ?MMPBSA.py is using cpptraj and mmpbsa_py_energy from Amber 12 it seems.
> ?
>
>
> > Preparing trajectories for simulation...
> > Usage: /usr/local/packages/amber12/amber12/bin/cpptraj [-p <Top1>, -p
> > <Top2>, ...] [-i <Input>] [-debug <N>]
> > /usr/local/packages/amber12/amber12/bin/cpptraj <Top1> <Input>
> > Additional options:
> > --help, -help: print usage information and exit.
> > -V, --version: print version information and exit.
> > --defines: print list of defines used in compilation.
> >
>
> ?This error output suggests that MMPBSA.py is feeding incorrect
> command-line arguments to cpptraj.
> ?
>
> > File "/usr/local/packages/amber14/bin/MMPBSA.py", line 95, in <module>
> > app.file_setup()
> >
>
> ?These traceback messages show that MMPBSA.py from Amber 14 is being used.
>
> This is likely your problem. cpptraj has probably changed between Amber 12
> and Amber 14, and MMPBSA.py uses features not present in earlier versions.
>
> In general, you can't mix-and-match different components of different
> AmberTools installations. You need to clean up your path to make sure that
> MMPBSA.py is finding the right AmberTools components (in particular, make
> sure AMBERHOME points to your latest Amber installation).
>
> I also recommend updating to the latest version of AmberTools.
>
> HTH,
> Jason
>
> --
> Jason M. Swails
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
------------------------------
Message: 6
Date: Tue, 4 Oct 2016 08:25:47 +0000 (UTC)
From: Maryam Hamzehee <maryam_h_7860.yahoo.com>
Subject: Re: [AMBER] MM-PBSA result for small molecule-receptor
complex
To: AMBER Mailing List <amber.ambermd.org>
Message-ID: <1416189448.348477.1475569547078.mail.yahoo.com>
Content-Type: text/plain; charset=UTF-8
Hi
Many thanks for the reply. Regarding the preparation of files for my small ligand I followed the tutorial4b for generation of small molecule parameters using antechamber (http://ambermd.org/tutorials/basic/tutorial4b/), I created the *.prmtop and *.inpcrd files for my ligand. However, by using the MM-PBSA calculation (tutorial A3), the result was surprising. The binding free energy for the complex of small ligand -receptor was around ~-4000kCal/mol based on GB calculations. Should I use ante-MMPBSA.py for generating of the ligand, receptor, and complex from the solvated form of complex??
ThanksMaryam
?
On Monday, 3 October 2016, 20:37, Bruno Falcone <brunofalcone.qo.fcen.uba.ar> wrote:
Hi, adding to what David has said, it sounds like you're probably
breaking bonds when defining the ligand. How did you generate the
parameter files? I recommend using ante-MMPBSA.py
Hope this helps,
Bruno
On 03/10/16 08:44, David A Case wrote:
> On Mon, Oct 03, 2016, Maryam Hamzehee wrote:
>
>> I am trying to do MM-PBSA calculation for determining binding energy for
>> the complex of receptor and an small ligand. I went through the MMBPSA
>> tutorial and used python-based version of MM-PBSA. Unfortunately PB did
>> not work, I ignored the PB and continued with GB. After calculation the
>> delta G was -4124.0239.
>> GENERALIZED BORN:
>> WARNING: INCONSISTENCIES EXIST WITHIN INTERNAL POTENTIALTERMS. THE
>> VALIDITY OF THESE RESULTS ARE HIGHLY QUESTIONABLE
> Please read the opening paragraphs in Chapter 31 of the Manual.? You need to
> be able to these calculations "by hand" first.? This is (fairly) easy with GB,
> since the averages of all the energy terms are printed in the mdout file, and
> you just need to subtract the receptor and ligand energies from that of the
> complex.? This will give you results that you can compare to the results of
> the python script, to try to see where the problems are coming from.
>
> (Above ignores contributions from configurational entropy; but you need to
> tackle the big problems first.)
>
> ...good luck....dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
------------------------------
Message: 7
Date: Tue, 4 Oct 2016 19:38:12 +0800
From: "xwnail2003.163.com" <xwnail2003.163.com>
Subject: [AMBER] did not find amber.python
To: amber <amber.ambermd.org>
Message-ID: <201610041938110172672.163.com>
Content-Type: text/plain; charset="GB2312"
Dear sir,
I am Wei Xu, coming from Jinan University, Guangzhou 510632, China. I am interesting in molecular dynamic simulation. In the stall, i put the command ? make install?, but the outcome is wrong. The feedback is that ?/home/xuwei/amber16/bin/amber.python: not found?.
I wanted to know how to solve the problem?
Sincerely yours,
Wei Xu
Dr. Xu,
------------------------------
Message: 8
Date: Tue, 4 Oct 2016 04:54:19 -0700
From: Bill Ross <ross.cgl.ucsf.edu>
Subject: Re: [AMBER] did not find amber.python
To: AMBER Mailing List <amber.ambermd.org>
Message-ID: <065c6866-bf94-7ee7-60d9-89a7452362c9.cgl.ucsf.edu>
Content-Type: text/plain; charset=gbk; format=flowed
Does the /home/xuwei/amber16/bin directory exist?
What instructions are you following? What operating system/version?
Bill
On 10/4/16 4:38 AM, xwnail2003.163.com wrote:
> Dear sir,
> I am Wei Xu, coming from Jinan University, Guangzhou 510632, China. I am interesting in molecular dynamic simulation. In the stall, i put the command ? make install?, but the outcome is wrong. The feedback is that ?/home/xuwei/amber16/bin/amber.python: not found?.
> I wanted to know how to solve the problem?
> Sincerely yours,
> Wei Xu
>
>
>
> Dr. Xu,
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
------------------------------
Message: 9
Date: Tue, 4 Oct 2016 08:17:07 -0400
From: David A Case <david.case.rutgers.edu>
Subject: Re: [AMBER] MM-PBSA result for small molecule-receptor
complex
To: Maryam Hamzehee <maryam_h_7860.yahoo.com>, AMBER Mailing List
<amber.ambermd.org>
Message-ID: <20161004121706.GD77773.scarletmail.rutgers.edu>
Content-Type: text/plain; charset=iso-8859-1
On Tue, Oct 04, 2016, Maryam Hamzehee wrote:
> However, by using the MM-PBSA
> calculation (tutorial A3), the result was surprising. The binding
> free energy for the complex of small ligand -receptor was around
> ~-4000kCal/mol based on GB calculations. Should I use ante-MMPBSA.py for
> generating of the ligand, receptor, and complex from the solvated form
> of complex??
(I usually try to avoid repeating previous advice, but will break that rule
here: the following is aimed at everyone on the list, not just at this
problem.)
Free energy calculations are an advanced subject, and require diligence
and experience to carry out correctly. Scripts like MMPBSA.py or
ante-MMPBSA.py are useful tools for experienced users to automate the
calcualtions, but they are complex, and cannot possibly trap all errors.
Running MM/GB-SA calculations "by hand" (without the use of any of these
scripts) is not all that difficult: set up three GB trajectories (ligand,
receptor and complex); collect the average energies (in various categories,
e.g. bonds, angles, etc.) from each the three simulations; do the appropriate
subtractions. Use VMD or Chimera to visualize each of the trajectories to
make sure that they are doing reasonable things.
Once you do this, you should have a set of sample results that you fully
understand. If you get nonsensical binding energies, see if you can narrow
down the problem. By all means, post here if you get stuck. If you get
reasonable results that you understand, then you can re-try the automated
scripts--you will have a much better chance of finding out what is going
wrong.
It is certainly possible (even likely) that using ante-MMPBSA.py
will fix the problems you have encountered. If that happens, you can set
aside the "by hand" sample calculations that you did. But I strongly
recommend avoiding the automated scripts at the very beginning--after all,
I wrote the paragraphs at the beginning of Chap. 31 that make the same
point.
...good luck....dac
------------------------------
Message: 10
Date: Tue, 4 Oct 2016 14:48:27 +0200
From: Hirdesh Kumar <hirdesh.iitd.gmail.com>
Subject: [AMBER] time step changes during long simulation run
To: AMBER Mailing List <amber.ambermd.org>
Message-ID:
<CAPKknGp6D4M6GBofJgCFTZtwEOujU3K9Lw_qU+SoCMUrgYihjQ.mail.gmail.com>
Content-Type: text/plain; charset=UTF-8
Hi,
I am doing a long simulation of my protein-ligand complex. In the out file,
the time step was written as:
TIME(PS) = 335980.000 (note XXX.000 format)..
NSTEP =139790000 *TIME(PS) = 335980.000* TEMP(K) = 298.18 PRESS
= 15.6
Etot = -115980.9003 EKtot = 27977.8633 EPtot =
-143958.7636
BOND = 1085.4143 ANGLE = 2911.4185 DIHED =
4565.4169
1-4 NB = 1346.0456 1-4 EEL = 14330.8565 VDWAALS =
16001.0010
EELEC = -184198.9163 EHBOND = 0.0000 RESTRAINT =
0.0000
EKCMT = 12017.9146 VIRIAL = 11864.6655 VOLUME =
455008.3585
Density =
1.0337
------------------------------------------------------------------------------
wrapping first mol.: -27.96280 -39.54537 68.49461
NSTEP =139795000 *TIME(PS) = 335990.000* TEMP(K) = 299.93 PRESS =
-246.6
Etot = -116468.7242 EKtot = 28142.6172 EPtot =
-144611.3414
BOND = 1094.3586 ANGLE = 2907.2089 DIHED =
4540.0461
1-4 NB = 1310.6156 1-4 EEL = 14286.1251 VDWAALS =
16100.2964
EELEC = -184849.9921 EHBOND = 0.0000 RESTRAINT =
0.0000
EKCMT = 12061.4866 VIRIAL = 14481.3696 VOLUME =
454452.8877
Density =
1.0350
------------------------------------------------------------------------------
wrapping first mol.: -27.97826 -39.56723 68.53246
wrapping first mol.: -27.97826 -39.56723 68.53246
NSTEP =139800000 *TIME(PS) = 335999.999 * TEMP(K) = 297.37 PRESS
= 2.1
Etot = -116224.6694 EKtot = 27902.1836 EPtot =
-144126.8530
BOND = 1077.4677 ANGLE = 2884.7374 DIHED =
4519.3357
1-4 NB = 1305.2631 1-4 EEL = 14371.2153 VDWAALS =
15909.1364
EELEC = -184194.0086 EHBOND = 0.0000 RESTRAINT =
0.0000
EKCMT = 11897.3296 VIRIAL = 11876.4293 VOLUME =
455201.7252
Density =
1.0333
------------------------------------------------------------------------------
wrapping first mol.: -27.98357 -39.57475 68.54548
As you can see the output was saved at every 10ps. And after 335990.000
ps, the expected next line should be:
TIME(PS) = 330000.000 PS. But, it was written as TIME(PS) = 335999.999..
This point onward, the output is always in ***.999 format, which is new to
me. I checked the trajectories and the system looks fine.
Is it the normal behaviour or there is something wrong ?
Thanks,
Hirdesh
------------------------------
Message: 11
Date: Tue, 4 Oct 2016 09:21:55 -0400
From: Jason Swails <jason.swails.gmail.com>
Subject: Re: [AMBER] time step changes during long simulation run
To: AMBER Mailing List <amber.ambermd.org>
Message-ID:
<CAEk9e3pVKHKgRqYkin8_sGdezVJ0u6X7qmD+EbbWF25XvXTzoQ.mail.gmail.com>
Content-Type: text/plain; charset=UTF-8
On Tue, Oct 4, 2016 at 8:48 AM, Hirdesh Kumar <hirdesh.iitd.gmail.com>
wrote:
> Hi,
> I am doing a long simulation of my protein-ligand complex. In the out file,
> the time step was written as:
> TIME(PS) = 335980.000 (note XXX.000 format)..
>
>
> NSTEP =139790000 *TIME(PS) = 335980.000* TEMP(K) = 298.18 PRESS
> = 15.6
> Etot = -115980.9003 EKtot = 27977.8633 EPtot =
> -143958.7636
> BOND = 1085.4143 ANGLE = 2911.4185 DIHED =
> 4565.4169
> 1-4 NB = 1346.0456 1-4 EEL = 14330.8565 VDWAALS =
> 16001.0010
> EELEC = -184198.9163 EHBOND = 0.0000 RESTRAINT =
> 0.0000
> EKCMT = 12017.9146 VIRIAL = 11864.6655 VOLUME =
> 455008.3585
> Density =
> 1.0337
> ------------------------------------------------------------
> ------------------
>
> wrapping first mol.: -27.96280 -39.54537 68.49461
>
> NSTEP =139795000 *TIME(PS) = 335990.000* TEMP(K) = 299.93 PRESS =
> -246.6
> Etot = -116468.7242 EKtot = 28142.6172 EPtot =
> -144611.3414
> BOND = 1094.3586 ANGLE = 2907.2089 DIHED =
> 4540.0461
> 1-4 NB = 1310.6156 1-4 EEL = 14286.1251 VDWAALS =
> 16100.2964
> EELEC = -184849.9921 EHBOND = 0.0000 RESTRAINT =
> 0.0000
> EKCMT = 12061.4866 VIRIAL = 14481.3696 VOLUME =
> 454452.8877
> Density =
> 1.0350
> ------------------------------------------------------------
> ------------------
>
> wrapping first mol.: -27.97826 -39.56723 68.53246
> wrapping first mol.: -27.97826 -39.56723 68.53246
>
> NSTEP =139800000 *TIME(PS) = 335999.999 * TEMP(K) = 297.37 PRESS
> = 2.1
> Etot = -116224.6694 EKtot = 27902.1836 EPtot =
> -144126.8530
> BOND = 1077.4677 ANGLE = 2884.7374 DIHED =
> 4519.3357
> 1-4 NB = 1305.2631 1-4 EEL = 14371.2153 VDWAALS =
> 15909.1364
> EELEC = -184194.0086 EHBOND = 0.0000 RESTRAINT =
> 0.0000
> EKCMT = 11897.3296 VIRIAL = 11876.4293 VOLUME =
> 455201.7252
> Density =
> 1.0333
> ------------------------------------------------------------
> ------------------
>
> wrapping first mol.: -27.98357 -39.57475 68.54548
>
>
>
>
> As you can see the output was saved at every 10ps. And after 335990.000
> ps, the expected next line should be:
>
> TIME(PS) = 330000.000 PS. But, it was written as TIME(PS) = 335999.999..
>
> This point onward, the output is always in ***.999 format, which is new to
> me. I checked the trajectories and the system looks fine.
>
> Is it the normal behaviour or there is something wrong ?
>
?This is normal. It is basic round-off error. The primary time unit is
picoseconds, but your time-step is on the order of femtoseconds -- 0.001 or
0.002 ps. However, both 0.001 and 0.002 are repeating decimals in binary
(which is how computers store numbers), which means that those two numbers
are *impossible* to represent exactly by a simple floating point variable.?
So to the computer, the time step is *very close* to 0.001 or 0.002
(whichever you chose), but not exactly the same. If you add that
difference up 330,000,000 times (or 165,000,000 times for a 2 fs time
step), you get something close to 0.001.
?At that point, the difference appears in the representation of the number
in the output file.
HTH,?
Jason
P.S., you can demonstrate this with your own program. Here is a C program
that illustrates exactly what's happening:
#include <stdio.h>
int main() {
const double dt = 0.002;
double t = 0;
long long int i;
for (i = 0; i < 165000000; i++) {
t += dt;
}
printf("t = %.4f\n", t);
return 0;
}
When I compile this program ("gcc test.c") and run the resulting a.out file
("./a.out"), I see the following output:
$ gcc test.c
$ ./a.out
t = 329999.9996
--
Jason M. Swails
------------------------------
Message: 12
Date: Tue, 4 Oct 2016 15:26:40 +0200
From: Hirdesh Kumar <hirdesh.iitd.gmail.com>
Subject: Re: [AMBER] time step changes during long simulation run
To: AMBER Mailing List <amber.ambermd.org>
Message-ID:
<CAPKknGogK6xQxde7No+jsUnkC2XYi=33ieptVH41o7B3TVW44Q.mail.gmail.com>
Content-Type: text/plain; charset=UTF-8
Thanks Jason,
?Best,
Hirdesh?
On Tue, Oct 4, 2016 at 3:21 PM, Jason Swails <jason.swails.gmail.com> wrote:
> On Tue, Oct 4, 2016 at 8:48 AM, Hirdesh Kumar <hirdesh.iitd.gmail.com>
> wrote:
>
> > Hi,
> > I am doing a long simulation of my protein-ligand complex. In the out
> file,
> > the time step was written as:
> > TIME(PS) = 335980.000 (note XXX.000 format)..
> >
> >
> > NSTEP =139790000 *TIME(PS) = 335980.000* TEMP(K) = 298.18 PRESS
> > = 15.6
> > Etot = -115980.9003 EKtot = 27977.8633 EPtot =
> > -143958.7636
> > BOND = 1085.4143 ANGLE = 2911.4185 DIHED =
> > 4565.4169
> > 1-4 NB = 1346.0456 1-4 EEL = 14330.8565 VDWAALS =
> > 16001.0010
> > EELEC = -184198.9163 EHBOND = 0.0000 RESTRAINT =
> > 0.0000
> > EKCMT = 12017.9146 VIRIAL = 11864.6655 VOLUME =
> > 455008.3585
> > Density =
> > 1.0337
> > ------------------------------------------------------------
> > ------------------
> >
> > wrapping first mol.: -27.96280 -39.54537 68.49461
> >
> > NSTEP =139795000 *TIME(PS) = 335990.000* TEMP(K) = 299.93 PRESS =
> > -246.6
> > Etot = -116468.7242 EKtot = 28142.6172 EPtot =
> > -144611.3414
> > BOND = 1094.3586 ANGLE = 2907.2089 DIHED =
> > 4540.0461
> > 1-4 NB = 1310.6156 1-4 EEL = 14286.1251 VDWAALS =
> > 16100.2964
> > EELEC = -184849.9921 EHBOND = 0.0000 RESTRAINT =
> > 0.0000
> > EKCMT = 12061.4866 VIRIAL = 14481.3696 VOLUME =
> > 454452.8877
> > Density =
> > 1.0350
> > ------------------------------------------------------------
> > ------------------
> >
> > wrapping first mol.: -27.97826 -39.56723 68.53246
> > wrapping first mol.: -27.97826 -39.56723 68.53246
> >
> > NSTEP =139800000 *TIME(PS) = 335999.999 * TEMP(K) = 297.37 PRESS
> > = 2.1
> > Etot = -116224.6694 EKtot = 27902.1836 EPtot =
> > -144126.8530
> > BOND = 1077.4677 ANGLE = 2884.7374 DIHED =
> > 4519.3357
> > 1-4 NB = 1305.2631 1-4 EEL = 14371.2153 VDWAALS =
> > 15909.1364
> > EELEC = -184194.0086 EHBOND = 0.0000 RESTRAINT =
> > 0.0000
> > EKCMT = 11897.3296 VIRIAL = 11876.4293 VOLUME =
> > 455201.7252
> > Density =
> > 1.0333
> > ------------------------------------------------------------
> > ------------------
> >
> > wrapping first mol.: -27.98357 -39.57475 68.54548
> >
> >
> >
> >
> > As you can see the output was saved at every 10ps. And after 335990.000
> > ps, the expected next line should be:
> >
> > TIME(PS) = 330000.000 PS. But, it was written as TIME(PS) =
> 335999.999..
> >
> > This point onward, the output is always in ***.999 format, which is new
> to
> > me. I checked the trajectories and the system looks fine.
> >
> > Is it the normal behaviour or there is something wrong ?
> >
>
> ?This is normal. It is basic round-off error. The primary time unit is
> picoseconds, but your time-step is on the order of femtoseconds -- 0.001 or
> 0.002 ps. However, both 0.001 and 0.002 are repeating decimals in binary
> (which is how computers store numbers), which means that those two numbers
> are *impossible* to represent exactly by a simple floating point variable.?
> So to the computer, the time step is *very close* to 0.001 or 0.002
> (whichever you chose), but not exactly the same. If you add that
> difference up 330,000,000 times (or 165,000,000 times for a 2 fs time
> step), you get something close to 0.001.
>
> ?At that point, the difference appears in the representation of the number
> in the output file.
>
> HTH,?
> Jason
>
> P.S., you can demonstrate this with your own program. Here is a C program
> that illustrates exactly what's happening:
>
> #include <stdio.h>
>
> int main() {
> const double dt = 0.002;
> double t = 0;
> long long int i;
>
> for (i = 0; i < 165000000; i++) {
> t += dt;
> }
> printf("t = %.4f\n", t);
>
> return 0;
> }
>
> When I compile this program ("gcc test.c") and run the resulting a.out file
> ("./a.out"), I see the following output:
>
> $ gcc test.c
> $ ./a.out
> t = 329999.9996
>
> --
> Jason M. Swails
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
------------------------------
Message: 13
Date: Tue, 4 Oct 2016 10:58:31 -0300
From: Bruno Falcone <brunofalcone.qo.fcen.uba.ar>
Subject: Re: [AMBER] MM-PBSA result for small molecule-receptor
complex
To: david.case.rutgers.edu, AMBER Mailing List <amber.ambermd.org>
Message-ID: <4fe8c65c-0ead-f128-6c3e-9211550c5d3c.qo.fcen.uba.ar>
Content-Type: text/plain; charset=windows-1252; format=flowed
Hi Maryam,
If you're keen to share the complex parameter and 10 frames from your MD
with stripped solvent I can have a quick look to see if I can spot any
errors.
Cheers,
Bruno
On 04/10/16 09:17, David A Case wrote:
> On Tue, Oct 04, 2016, Maryam Hamzehee wrote:
>
>> However, by using the MM-PBSA
>> calculation (tutorial A3), the result was surprising. The binding
>> free energy for the complex of small ligand -receptor was around
>> ~-4000kCal/mol based on GB calculations. Should I use ante-MMPBSA.py for
>> generating of the ligand, receptor, and complex from the solvated form
>> of complex?
> (I usually try to avoid repeating previous advice, but will break that rule
> here: the following is aimed at everyone on the list, not just at this
> problem.)
>
> Free energy calculations are an advanced subject, and require diligence
> and experience to carry out correctly. Scripts like MMPBSA.py or
> ante-MMPBSA.py are useful tools for experienced users to automate the
> calcualtions, but they are complex, and cannot possibly trap all errors.
>
> Running MM/GB-SA calculations "by hand" (without the use of any of these
> scripts) is not all that difficult: set up three GB trajectories (ligand,
> receptor and complex); collect the average energies (in various categories,
> e.g. bonds, angles, etc.) from each the three simulations; do the appropriate
> subtractions. Use VMD or Chimera to visualize each of the trajectories to
> make sure that they are doing reasonable things.
>
> Once you do this, you should have a set of sample results that you fully
> understand. If you get nonsensical binding energies, see if you can narrow
> down the problem. By all means, post here if you get stuck. If you get
> reasonable results that you understand, then you can re-try the automated
> scripts--you will have a much better chance of finding out what is going
> wrong.
>
> It is certainly possible (even likely) that using ante-MMPBSA.py
> will fix the problems you have encountered. If that happens, you can set
> aside the "by hand" sample calculations that you did. But I strongly
> recommend avoiding the automated scripts at the very beginning--after all,
> I wrote the paragraphs at the beginning of Chap. 31 that make the same
> point.
>
> ...good luck....dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
------------------------------
Message: 14
Date: Tue, 4 Oct 2016 09:52:25 -0700
From: Atila Petrosian <atila.petrosian.gmail.com>
Subject: [AMBER] MD simulation by amber
To: amber <amber.ambermd.org>
Message-ID:
<CAON_0oUtX1uvgSjdZS10mBQSoDMW0UzPBNjQjio3RXqcwpLxTg.mail.gmail.com>
Content-Type: text/plain; charset=UTF-8
Dear amber users,
Hi, I want to do molecular dynamic simulation with amber. The condition of
my protein-ligand system are following as: my protein has Zn+2 ion in the
active site which three histidine coordinate with it by covalent bond. Also
my ligands are sulfonamide and have -1 charge on nitrogen atom which are
oriented from nitrogen atom toward zinc ion in the active site of the
protein.
I wonder if you recommend me a suitable amber tutorial for such kind of
system I explain.
Regards,
Yeganeh
------------------------------
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
End of AMBER Digest, Vol 1717, Issue 1
**************************************
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: InsertPic_.png)
