Date: Thu, 8 Sep 2016 20:35:38 +0530 (IST)
Hi,
I am running prot+ligand simulation for a couple of systems for understanding
ligand bound conformational changes. (I have used standard Amber14 protocols for
setting up the system, minimization, equilibration, etc.)
I have generated 100ns production run data and am using cpptraj version 14.25
for the analysis.
I had earlier run rmsd calculations for all systems using the following script :
strip :WAT
strip :Na+
center :1-250 mass
autoimage
rms first out rmsd.dat :1-250.CA
atomicfluct out Per-res-fluct.dat :1-250 byres
atomicfluct out CA-fluct.dat .CA byatom
go
rmsd was below 2Å and system appeared stable.
I have to re-run rmsd again, and am facing some problems inspite of using same
crd files, same prmtop file and same rmsd.in file :
1) RMSD data shows RMS as high as 25-45Å for a selected number of snapshots
after an interval of 50 frames. I find this very weird since I used the same
data ( crd files) a few days back with the same prmtop file and with the same
cpptraj code, but did not see any of this earlier.
No errors were reported during cpptraj analysis. After data has been generated,
it looks something like.
When visualised in VMD, I do not see any drastic changes in the prot structure.
Everything appears to be OK.
Is there any way to rectify this ? What seems to be the problem? Is this an
imaging issue or have the trajectories become corrupted? If so, please suggest
remedial measures.
Thank you.
Regards,
B.K. Dhanasekaran
-- This message has been scanned for viruses and dangerous content by MailScanner, and is believed to be clean.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
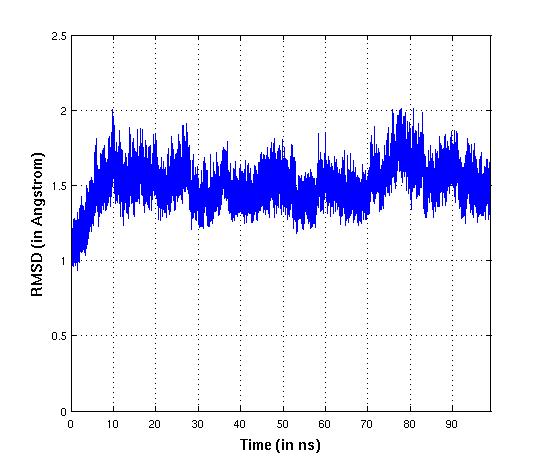
(image/jpeg attachment: old_rmsd_result.jpg)
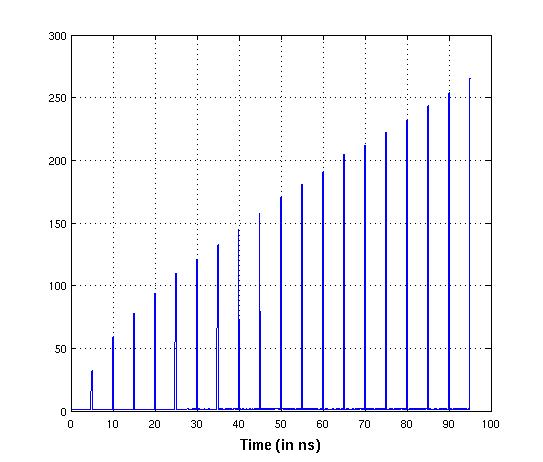
(image/jpeg attachment: new_rmsd_result.jpg)
