Date: Wed, 7 Sep 2016 12:16:23 -0300
Dear,
in the file .in that uses MCPB.py you have to specify
the program will use, as inputs are now created
to GAMESS and GAUSSIAN.
Have you done that? (I think it may be the problem, but have you ceterza
could show the error found)
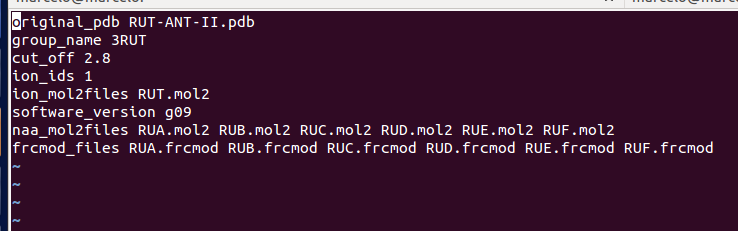
Here's a template that I used to GAUSSIAN:
[image: Imagem intercalada 1]
Regards,
Marcelo
Marcelo Andrade Chagas, MSc
(PhD student)
Laboratório de Química Computacional e Modelagem Molecular - LQC-MM
* http://lqcmm.qui.ufmg.br/
Departamento de Química da Universidade Federal de Minas Gerais - UFMG
Tel:(31)3409-5776
2016-09-07 6:15 GMT-03:00 Abhi Acharya <abhi117acharya.gmail.com>:
> Dear Amber Users,
>
> I am interested in deriving parameters for an active site containing Cu2+
> and Ca2+ ions. I am following the MCPB.py protocol (
> http://ambermd.org/tutorials/advanced/tutorial20/mcpbpy.htm).
>
> I have successfully performed geometry optimization and calculation of
> force constants for the optimized active site using GAMESS. However, it
> seems to me that one step after the geometry optimization and fc
> calculation is missing; the MCPB.py script asks for a gaussian fchk file
> which obviously will not be available if someone uses GAMESS. I am at loss
> about how to proceed hereon. I am hoping that someone here would suggest a
> method of using the GAMESS output for obtaining the amber parameters, using
> the MCPB.py workflow.
>
> Thank you.
> Abhishek Acharya
> Center for Cellular and Molecular Platforms
> NCBS, Bangalore.
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
