Date: Sat, 20 Aug 2016 18:13:41 +0000
Hi, Amy,
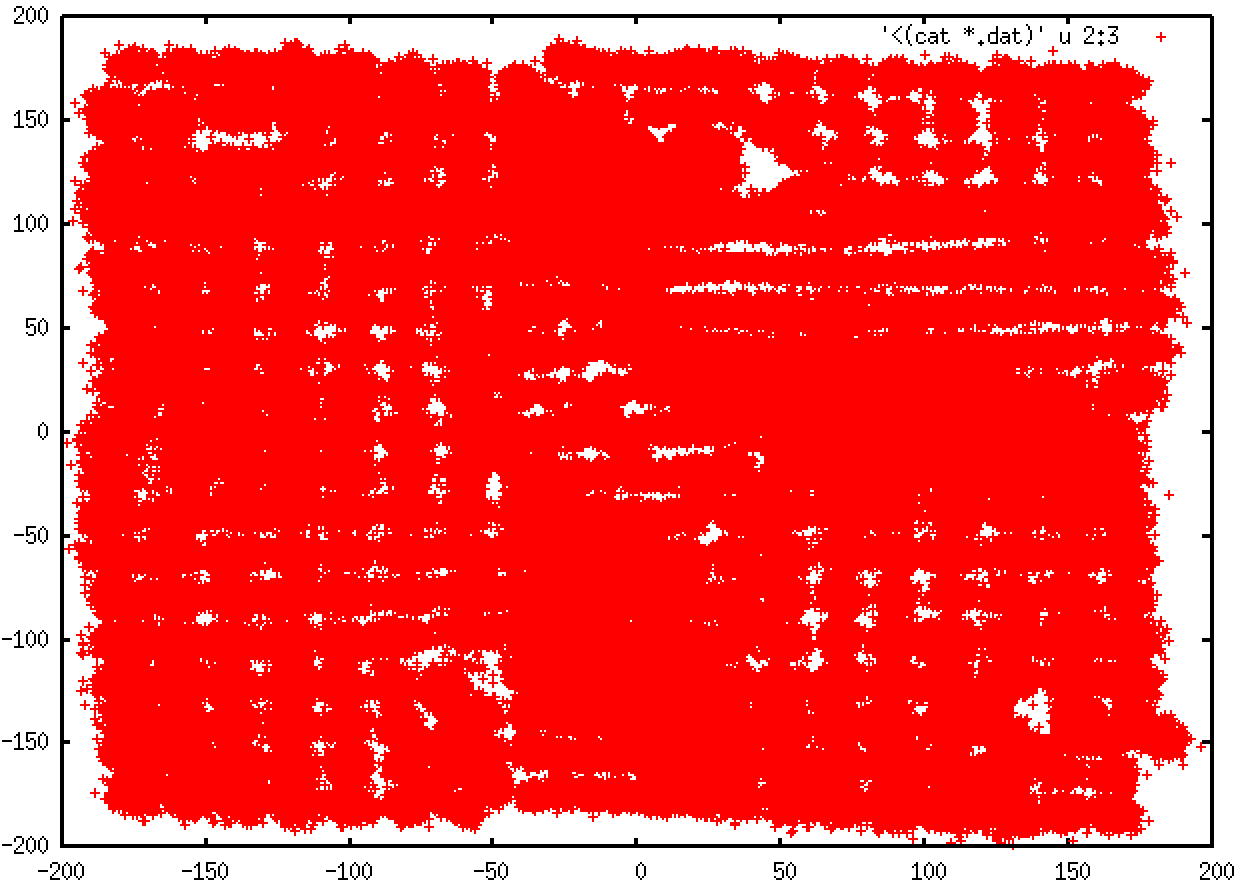
I guess there should not be "edges" in my PMF plot since my system is consist of two dihedrals and I turned on periodicity to both two dihedrals.
Attached is the overlapping of the windows along the two dihedrals along with the final results. When running this, I set all the force constants of all constraints in the metadata file to 0.06092 (meaning 100 kcal/mol/degree in Amber) just for test to avoid mistyping the force constants. The command line was:
./wham-2d Px -180 180 120 Py -180 180 120 0.0001 300 0 metadata result 0 >out
The final result looks weird. When I switched to use the metadata containing the real force constants, I got similar results.
Thank you for reply!
________________________________________
From: Amy Rice <arice3.hawk.iit.edu>
Sent: Saturday, August 20, 2016 11:41 AM
To: AMBER Mailing List
Subject: Re: [AMBER] WHAM analysis
Hi Abhishek,
There is not nearly enough information in your original email to let us
help you. Some information that would be useful to provide- What is your
reaction coordinate? How long is your simulation? Are you seeing sampling
in each bin?
Jin Chi- is the surface discontinuous when you plot it? It's hard to see
from the portion of the WHAM output that you provided, but since this is a
2D PMF, these infinite values could represent an edge and not necessarily a
discontinuity or hole in the PMF.
Hope this helps,
- Amy
On Sat, Aug 20, 2016 at 2:30 AM, Chi Jin <jinchi.chemistry.msu.edu> wrote:
> Me too. I ran wham-2d, the 4th column is the unnormalized probabilities:
>
> 118.500000 127.500000 7.185208 2.608360
> 118.500000 130.500000 6.997535 3.575855
> 118.500000 133.500000 7.204882 2.523505
> 118.500000 136.500000 6.304755 11.459057
> 118.500000 139.500000 inf 0.000000
> 118.500000 142.500000 inf 0.000000
> 118.500000 145.500000 inf 0.000000
> 118.500000 148.500000 6.228352 13.029487
> 118.500000 151.500000 6.640700 6.514633
> 118.500000 154.500000 5.443592 48.735702
> 118.500000 157.500000 inf 0.000000
> 118.500000 160.500000 4.994485 103.686285
>
> So that the PMF surface is not continuous, which doesn't sound reasonable
> to me.
> Jin Chi
> PhD student
> Department of Chemistry
> Michigan State University
> East Lansing, MI 48824
> Tel: (517)-355-9715 x261
> jinchi.chemistry.msu.edu
>
> ________________________________________
> From: Thakur, Abhishek <axt651.miami.edu>
> Sent: Friday, August 19, 2016 5:55 PM
> To: amber.ambermd.org
> Subject: [AMBER] WHAM analysis
>
> Hi
>
> I am trying to do WHAM analysis after QM/MM.
>
> But the result file that I am getting is [😎]
>
> #Coor Free
> 3.471739 0.221101
> 3.615217 inf
> 3.758696 inf
> 3.902174 inf
> 4.045652 inf
> 4.189130 1.645569
> 4.332609 0.000000
> 4.476087 inf
> 4.619565 inf
> 4.763043 inf
> 4.906522 inf
> 5.050000 2.616324
> 5.193478 0.022866
> 5.336957 2.745523
> 5.480435 inf
> 5.623913 inf
> 5.767391 inf
>
>
> Now I don't know why this inf is coming in my result file.
>
>
> Thanking you,
>
> Abhishek
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Amy Rice Ph.D. Student Physics Department Illinois Institute of Technology _______________________________________________ AMBER mailing list AMBER.ambermd.org http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 2D_WHAM.png)
- text/plain attachment: result.txt
